Clear Sky Science · en
Mithramycin alters EWS::FLI1 DNA binding and RNA polymerase II processivity to inhibit nascent transcription
Why this matters for cancer treatment
Ewing sarcoma is an aggressive bone cancer that strikes mainly children and young adults. It is driven by a powerful fusion protein, EWS::FLI1, that rewires how cells read their DNA. This study revisits an old chemotherapy drug, mithramycin, and shows that—if given in the right way—it can selectively shut down this cancer-driving program while sparing much of the rest of the cell’s machinery. The work reveals how dose and schedule, not just the drug itself, can make the difference between broad toxicity and precise targeting.
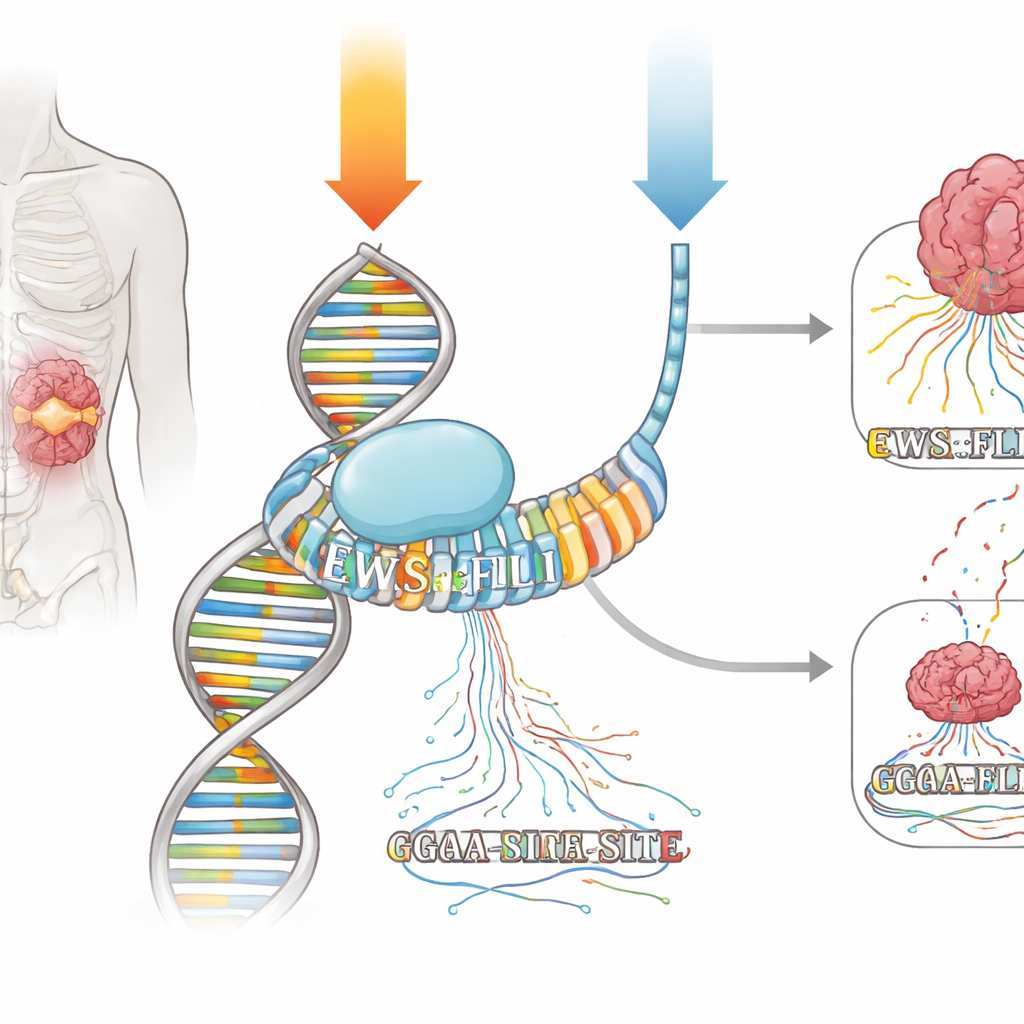
An old drug with a hidden talent
Mithramycin is a natural product that nestles into the minor groove of DNA, especially at short stretches rich in the letters G and C. For decades it has been labeled a blunt “chemotherapy” tool, assumed to damage DNA and block transcription across the board. However, doctors in the 1960s reported a complete response in a patient with Ewing sarcoma when mithramycin was given as a continuous seven-day infusion. More recently, when the same drug was tested again but delivered as short daily doses, only minimal benefit was seen. The authors set out to understand, with modern genomic tools, whether mithramycin can act as a precision drug against EWS::FLI1—and whether its schedule of administration explains the striking difference in patient responses.
How the fusion driver is switched on and off
EWS::FLI1 belongs to the ETS family of transcription factors, which latch onto a simple DNA word built around “GGAA.” In Ewing sarcoma, EWS::FLI1 binds especially strongly to long runs of repeated GGAA sites, called microsatellites, to turn whole networks of genes up or down. The team exposed Ewing sarcoma cells to two mithramycin regimens that produced similar suppression of key EWS::FLI1 target genes: a brief, high-dose pulse and a lower dose given for a much longer time. Using techniques that map both where EWS::FLI1 sits on DNA and where new RNA molecules are being made in real time, they could watch how each regimen changed binding and transcription across the genome.
Fine-tuning the transcription machinery
Contrary to older in vitro studies, mithramycin did not simply act as a universal transcription blocker at clinically relevant levels. At carefully chosen low concentrations, continuous exposure mainly altered how EWS::FLI1 starts transcription at its target sites. Some EWS::FLI1 peaks on DNA became weaker, others stronger, but in both cases the downstream effect was to “poison” the program: genes normally switched on by the fusion were turned down, and those normally suppressed were released. At higher concentrations, however, mithramycin began to interfere with the ability of RNA polymerase II—the enzyme that copies DNA into RNA—to travel along long genes. That stalling created broad, non-specific effects, masking some of the desirable reversal of EWS::FLI1’s activity, particularly for large genes that needed polymerase to move far from the start to the end of the gene.
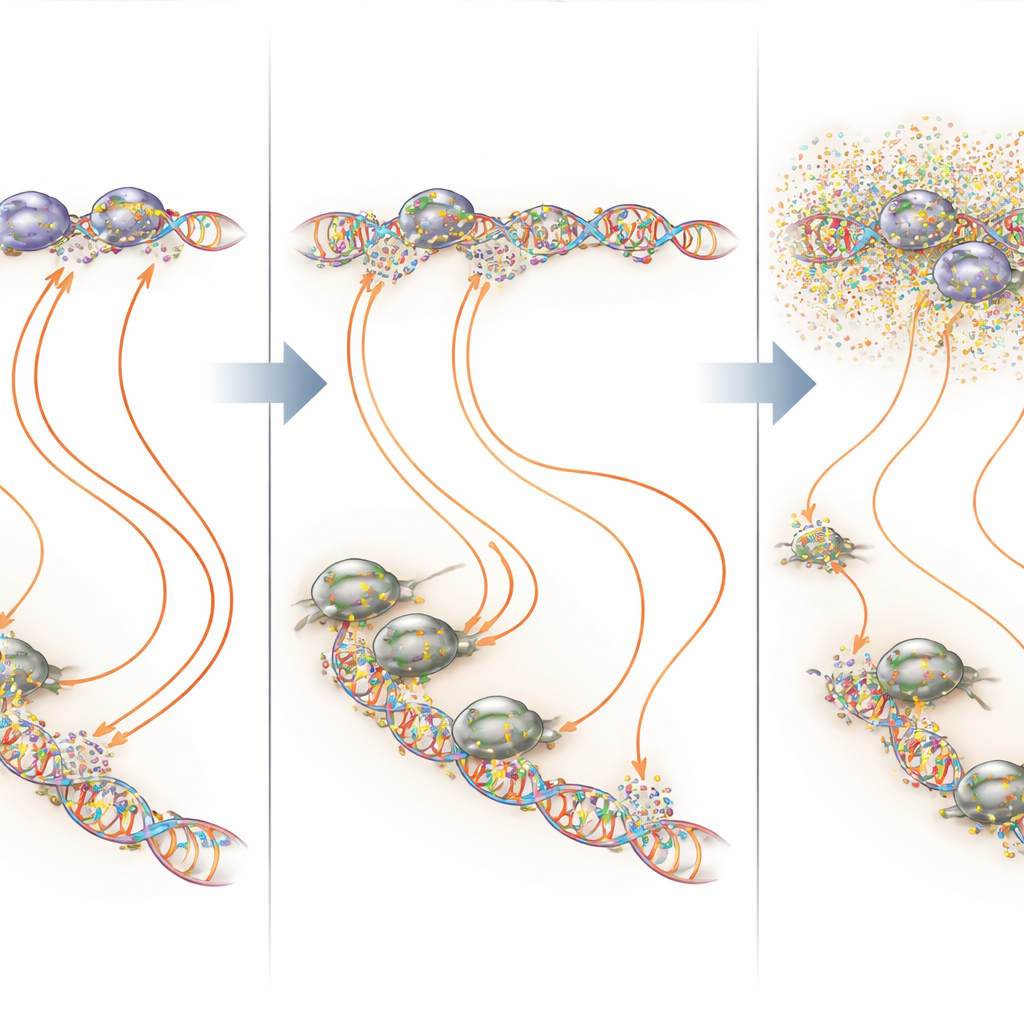
Targeting DNA hot spots instead of the whole genome
By overlaying binding and transcription maps, the researchers showed that mithramycin acts most strongly at the very GGAA-rich regions that EWS::FLI1 depends on. At these microsatellites and at isolated GGAA sites, low-dose continuous mithramycin often increased EWS::FLI1 occupancy but still shut down productive transcription—a clear illustration of a “Goldilocks” principle, where both too little and too much binding of a transcription factor can be harmful to its function. At other sites where EWS::FLI1 partners with cofactors such as AP-1, E2F, or RUNX2, mithramycin tended to destabilize the complexes and reduce binding. In all these scenarios, the net result was a more normal pattern of gene activity in the cancer cells, with much less disruption of unrelated genes when the drug was kept at a lower, steady level.
From lab mechanism to better treatment strategy
The team then tested whether this mechanistic insight mattered in living animals. In mice bearing Ewing sarcoma tumors, the same total dose of mithramycin given as a seven-day continuous infusion caused dramatic tumor shrinkage, whereas the same amount delivered as daily injections was far less effective. Importantly, this powerful effect occurred without signs of extra DNA damage in the tumors, arguing that the benefit comes from reprogramming transcription rather than injuring the genome. A newer, less toxic mithramycin analog, AIT-102, produced even more striking and durable tumor regressions when given by continuous infusion at relatively modest doses.
What this means for future cancer therapies
Overall, the study reveals mithramycin as a prototype “transcription factor modulator” rather than a crude transcription poison. By binding selectively to DNA sequences used by EWS::FLI1 and related ETS factors, it can dismantle a fusion-driven gene network that is otherwise very difficult to drug. Crucially, this specificity emerges only within a narrow window of dose and schedule, where continuous low-level exposure blocks the oncogenic program without broadly stalling the cell’s transcription machinery. These findings argue that, for drugs targeting transcription and chromatin, how we give the drug can be as important as the molecule itself, and they support renewed clinical development of mithramycin analogs for Ewing sarcoma and other cancers that rely on ETS transcription factors.
Citation: Kaufman, R., Flores, G., Boguslawski, E.A. et al. Mithramycin alters EWS::FLI1 DNA binding and RNA polymerase II processivity to inhibit nascent transcription. Nat Commun 17, 2844 (2026). https://doi.org/10.1038/s41467-026-69488-9
Keywords: Ewing sarcoma, mithramycin, EWS::FLI1, transcription factor targeting, drug dosing schedule