Clear Sky Science · en
A specific stem cell program and CD112 immunological axis dysfunctions underpinning monosomy 7-associated myeloid neoplasms
Why this blood problem matters
Some aggressive blood cancers share a puzzling change: patients lose an entire copy of chromosome 7 in their bone marrow cells. This defect, called monosomy 7, is linked to poor outcomes, yet doctors still lack targeted treatments for it. This study explores what is happening inside these cells at the level of genes and immune signals, revealing how a special stem cell program and a faulty immune “brake-and-gas” system help these cancers grow and escape the body’s defenses.
A risky shift in bone marrow stem cells
Myelodysplastic syndromes and acute myeloid leukemia are diseases in which immature blood cells take over the bone marrow. The authors compared bone marrow samples from patients with monosomy 7 to samples from other leukemia patients and healthy donors. By reading out which genes were switched on or off, they found that monosomy 7 cells carried a distinct “stemness” program: a set of 49 genes that keep cells in a self-renewing, stem-like state instead of allowing them to mature into normal blood cells. Many of these genes had already been tied to worse survival or unusual energy use in blood cancers, suggesting that this stem-cell pattern helps explain why monosomy 7 neoplasms are so hard to treat.
Hidden chemical marks that steer gene activity
The team next looked at DNA methylation, a chemical tag that can silence or alter gene activity without changing the DNA letters themselves. Using a high-resolution method, they showed that monosomy 7 bone marrow had a characteristic methylation “fingerprint,” with many regions more heavily tagged than normal. These changes did not mainly sit at gene start sites, but instead accumulated in intergenic enhancers, control regions that act like distant dimmer switches. Many of these enhancers clustered on specific chromosomes, including the remaining copy of chromosome 7, hinting that the intact copy is being rewired rather than simply acting as a passive leftover.
When control switches for key genes are rewired
Because enhancers are docking sites for transcription factors, the proteins that turn genes on and off, the researchers asked which factors might be affected. They found that binding sites for several homeobox proteins, master regulators of development, were often hypermethylated. By combining these chemical maps with public datasets of homeobox protein binding, they identified a large group of “HOX target” genes that were abnormally active in monosomy 7 cases. Many of these overlapped with the 49-gene stemness program. Experiments confirmed that at least one of these regulators, PAX8, failed to bind certain enhancers when they were hypermethylated, supporting the idea that altered enhancer chemistry helps lock cells into a cancerous stem-like state.
A broken immune checkpoint in the bone marrow
The work then turned to IKZF1, a gene on chromosome 7 that helps organize the 3D structure of DNA and regulate other genes. With only one copy of chromosome 7, monosomy 7 cells produced less IKZF1 and showed a pattern of genes typical of “IKZF1 insufficiency.” Among these was CD112, a protein on the cell surface that talks to immune cells. The researchers proved that IKZF1 normally binds the CD112 promoter and keeps it in check. When they reduced IKZF1 in healthy stem cells using CRISPR, CD112 levels rose, mirroring what they saw in patients. In bone marrow tissue from people with monosomy 7, leukemia blasts and abnormal precursors were strongly positive for CD112, whereas normal samples showed only scattered weak staining.
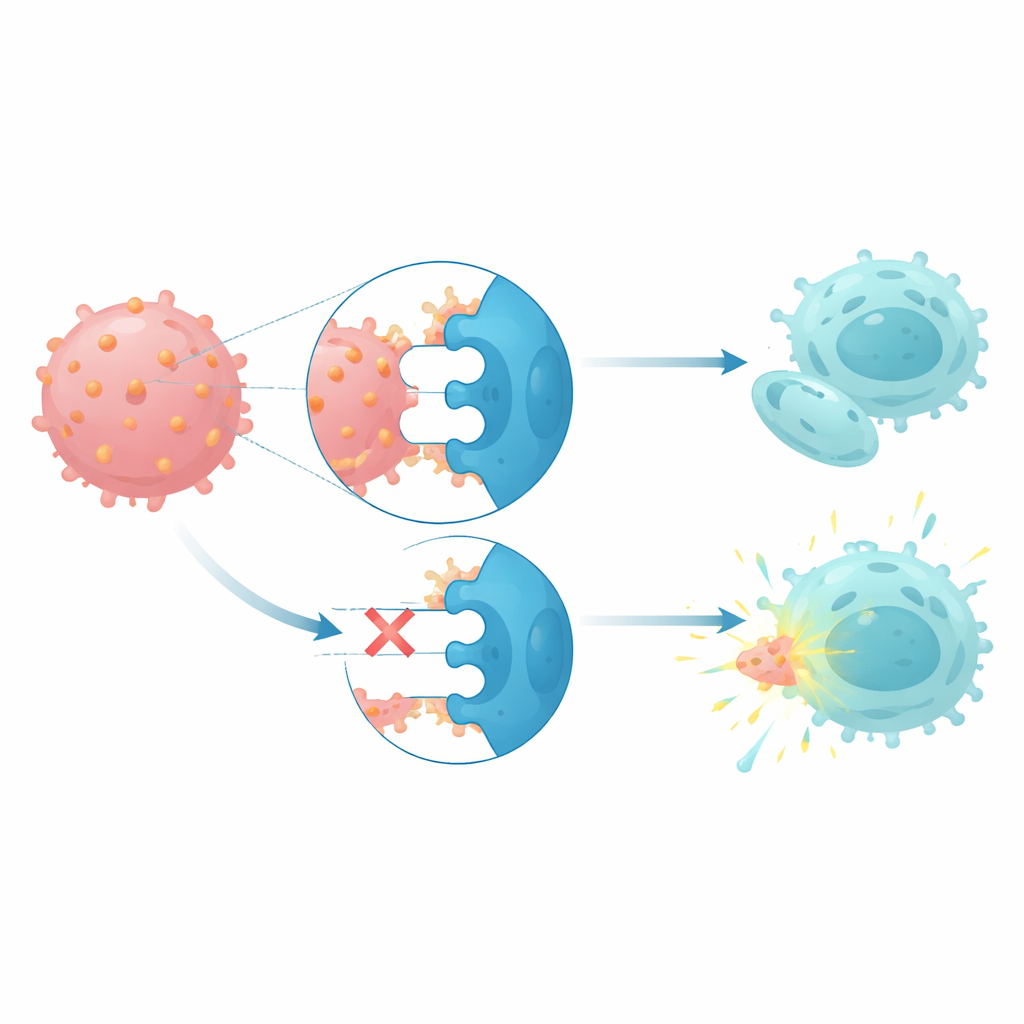
How leukemia cells disarm natural killer cells
CD112 interacts with three receptors on natural killer cells and T cells: an activating receptor called DNAM1 and two inhibitory receptors, TIGIT and PVRIG. In patients with monosomy 7, natural killer cells and T cells displayed higher levels of the inhibitory receptors and lower levels of the activating one, a pattern consistent with immune cells being pushed into a sluggish state. When healthy natural killer cells were co-cultured with CD112-high leukemia cells, their PVRIG levels rose and DNAM1 levels fell, suggesting that contact with these cancer cells helps reprogram immune defenses. Functional killing assays showed that blocking both TIGIT and PVRIG on patient natural killer cells restored their ability to lyse their own monosomy 7 leukemia blasts, while blocking CD112 alone was less effective.
What this could mean for future treatment
Together, these findings paint monosomy 7 myeloid neoplasms as cancers driven by a stem-like gene program and shielded by a distorted CD112-based immune checkpoint. By connecting loss of chromosome 7 to enhancer changes, altered master regulators, and a rewired CD112–TIGIT–PVRIG–DNAM1 axis, the study suggests that drugs blocking the inhibitory receptors TIGIT and PVRIG, possibly alongside existing DNA methylation therapies, could help patients’ own natural killer cells recognize and attack these otherwise hard-to-treat cancers.
Citation: Lema Fernandez, A.G., Nardelli, C., Quintini, M. et al. A specific stem cell program and CD112 immunological axis dysfunctions underpinning monosomy 7-associated myeloid neoplasms. Sig Transduct Target Ther 11, 173 (2026). https://doi.org/10.1038/s41392-026-02681-w
Keywords: monosomy 7, myeloid neoplasm, acute myeloid leukemia, immune checkpoint, natural killer cells