Clear Sky Science · en
Targeting GPR34 in damage-associated macrophages enhances anti-tumor immunity and the efficacy of Surufatinib in pancreatic cancer
Why this study matters
Pancreatic cancer is one of the deadliest cancers, in part because it is surrounded by an immune “force field” that shields the tumor from attack. This paper uncovers a key group of immune cells that help build that shield and shows that disabling a single molecular switch on those cells can make standard treatments work much better. For patients, this research points toward smarter drug combinations that could finally tip the balance in favor of the body’s own defenses.
The hidden helpers around the tumor
Pancreatic tumors grow inside a packed neighborhood of immune and connective cells known as the tumor microenvironment. Among the most influential residents are macrophages—white blood cells that normally clean up dead tissue and help coordinate repair. In pancreatic cancer, these tumor-associated macrophages often flip sides, dampening immune responses and helping the cancer resist treatment. A drug called Surufatinib, which is already used for other cancers, targets one of the main growth signals that keeps these macrophages alive. But its impact in pancreatic cancer has been modest, suggesting that macrophages may have backup routes that preserve their tumor-protective roles.
Following individual cells during treatment
The researchers launched a clinical trial in which people with pancreatic cancer received Surufatinib together with standard chemotherapy before surgery. Using single-cell RNA sequencing—a technique that reads gene activity in thousands of individual cells—they mapped all cell types in the removed tumors and compared patients who responded to treatment with those whose tumors kept growing. Non-responders had many more macrophages overall, and one subtype in particular dominated. These cells carried high levels of a receptor called GPR34 and showed gene programs linked to tissue damage responses, waste clearance, and creation of an immune-suppressive environment. At the same time, tumors from non-responders contained more worn-out, “exhausted” killer T cells that had lost their ability to efficiently attack cancer cells.
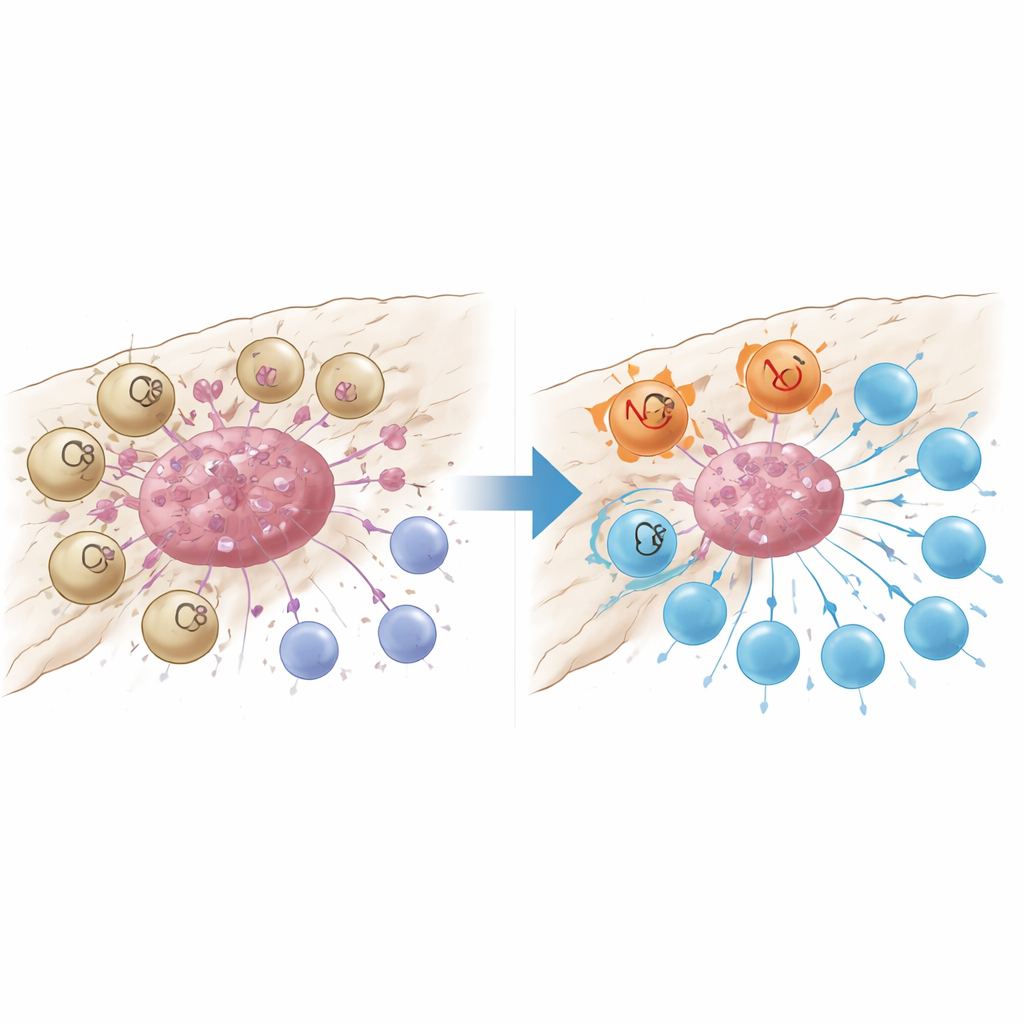
A damage sensor that turns off T cells
Closer analysis revealed that GPR34-marked macrophages were tightly clustered inside tumor nests and were rare in surrounding normal tissue or inflamed but noncancerous pancreas. Patients whose tumors contained more of these cells had fewer cancer-fighting CD8 T cells, more regulatory T cells that restrain immunity, and shorter survival times. In mouse models engineered to lack GPR34 only in macrophages, chemotherapy suddenly became much more effective: tumors shrank more, and T cells inside the tumor were more numerous and less exhausted. Laboratory co-culture experiments showed why. When macrophages with GPR34 sensed molecular “danger signals” released by dying tumor cells—especially a lipid called lysophosphatidylserine—they ramped up a process called efferocytosis, in which they engulf dead cell fragments. This not only increased their ability to swallow tumor debris but also triggered internal lysosomal pathways that broke down key molecules they need to show tumor antigens to T cells.
Two ways to silence immune attack
GPR34-driven macrophages undermined T cells in a second way: by secreting high levels of the signaling protein CXCL16 after taking up damaged tumor material. CXCL16 acted as a chronic stimulation signal, pushing CD8 T cells toward an exhausted state where they express brakes on their surface and lose killing power. Blocking GPR34 reduced CXCL16 release, restored antigen display on macrophages, and revitalized T cell function. The team traced the chain of events from the damage signal lipid, through GPR34 and a common growth pathway inside cells, to activation of efferocytosis machinery and lysosomes. When they pharmacologically blocked the downstream engulfment or lysosomal steps, macrophages kept more of their antigen-displaying molecules and supported more potent T-cell responses, underscoring that overactive cleanup can paradoxically hide the tumor from immune surveillance.
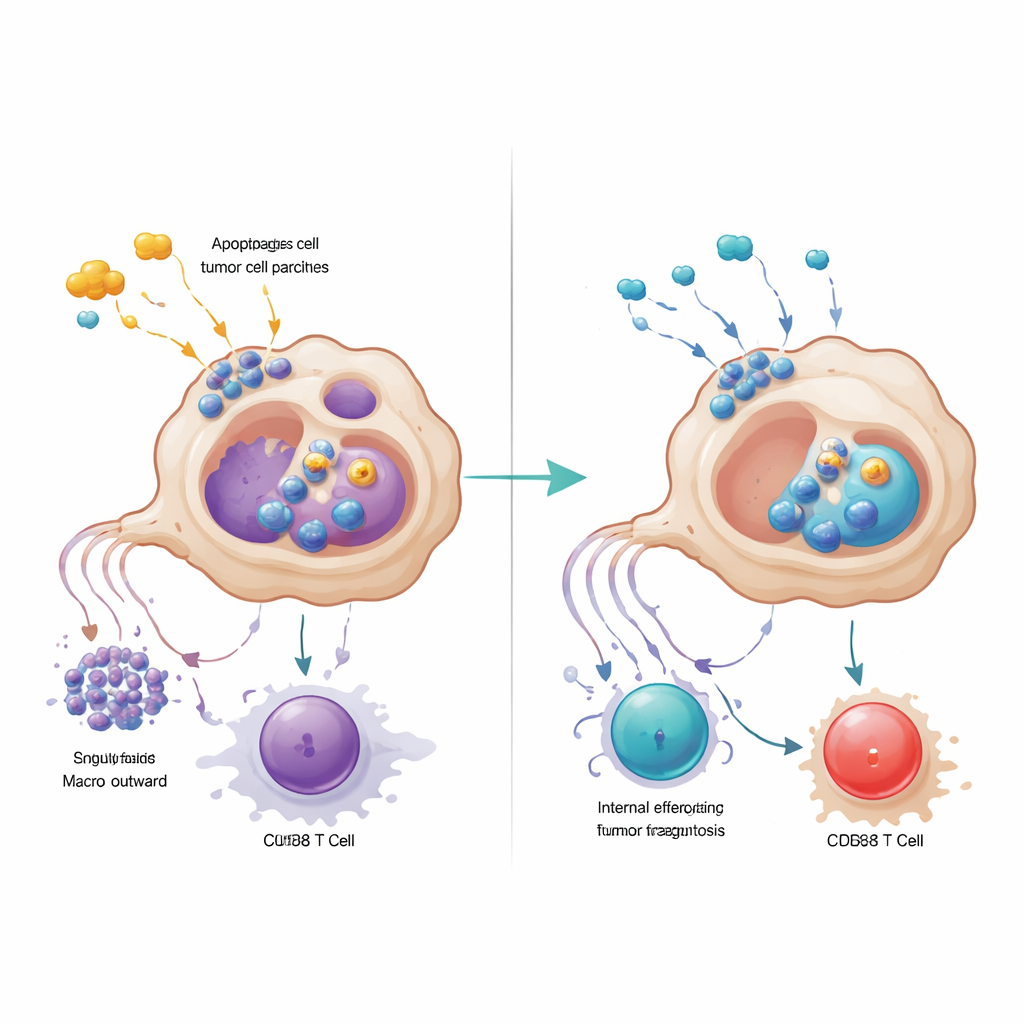
Making existing drugs work harder
Armed with this mechanistic map, the researchers tested a small-molecule GPR34 blocker in several mouse models and in patient-derived tumor organoids grown with immune cells. Adding the GPR34 antagonist to chemotherapy and Surufatinib consistently led to smaller tumors, more tumor cell death, stronger CD8 T-cell activity, and fewer signs of T-cell exhaustion, without obvious added toxicity in major organs or blood counts. In contrast, directly neutralizing CXCL16 alone did not reproduce these benefits, highlighting that the central leverage point is the macrophage damage sensor and its impact on both efferocytosis and antigen presentation. In plain terms, the study suggests that some macrophages in pancreatic cancer act like overzealous street sweepers that not only clear debris but also erase the clues the immune system needs. Turning off their GPR34 switch lets chemotherapy damage expose the tumor rather than hide it, opening the door to more durable and effective treatment combinations.
Citation: Guo, X., Liu, Y., Li, T. et al. Targeting GPR34 in damage-associated macrophages enhances anti-tumor immunity and the efficacy of Surufatinib in pancreatic cancer. Sig Transduct Target Ther 11, 156 (2026). https://doi.org/10.1038/s41392-026-02641-4
Keywords: pancreatic cancer, tumor microenvironment, macrophages, immunotherapy, Surufatinib