Clear Sky Science · en
Molecular mechanisms of NLRP3 inflammasome activation
Why Our Cells Light a Molecular Fire Alarm
Our bodies rely on frontline immune cells to notice trouble—invading microbes, toxic particles, or internal stress—and raise the alarm fast. One of the most powerful alarm switches is a protein machine called the NLRP3 inflammasome. When it flips on, it sparks inflammation that can save us from infection. But if this switch is stuck in the “on” position, it can drive a long list of diseases, from gout and diabetes to neurodegeneration. This review article explains, in molecular detail, how the NLRP3 inflammasome is built, what turns it on, and how cells fine‑tune its activity, offering clues for future drugs that cool harmful inflammation without silencing useful defenses.
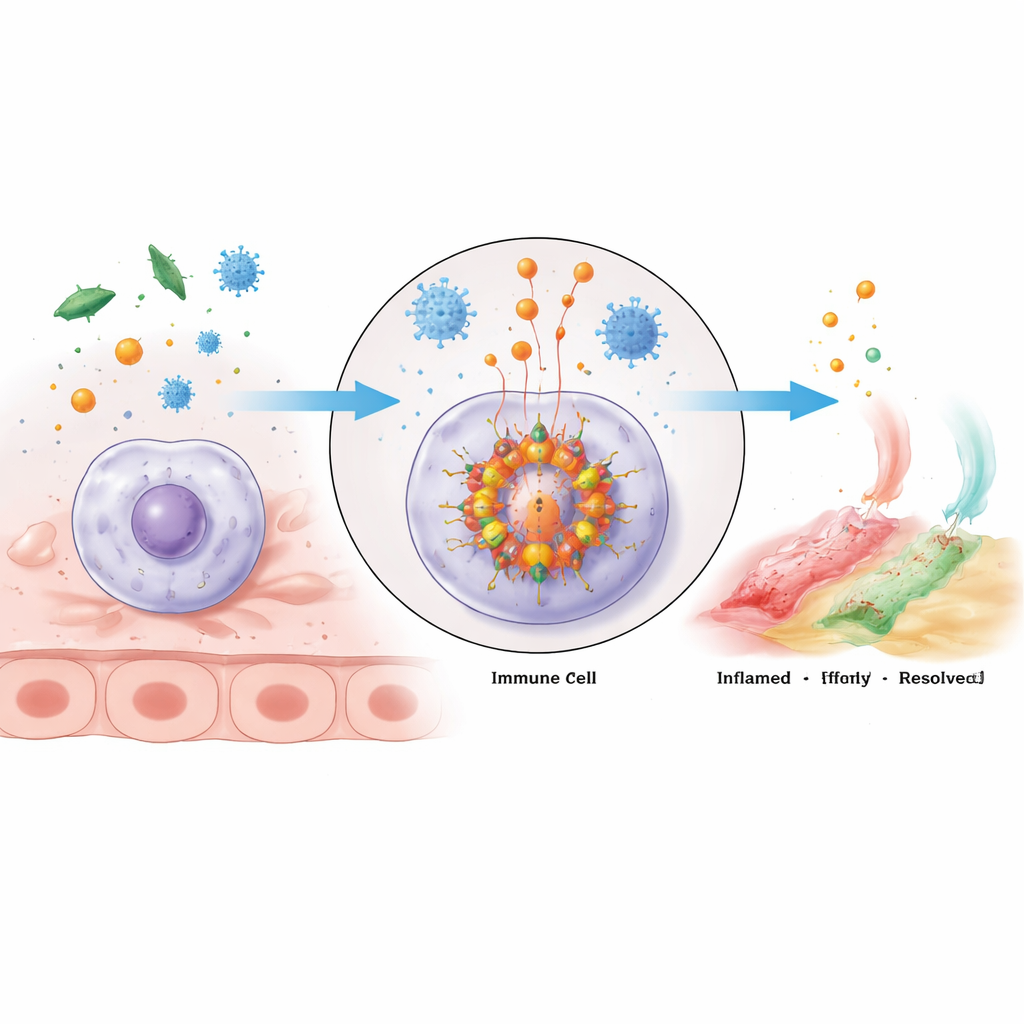
A Molecular Alarm Cluster Inside Immune Cells
The NLRP3 inflammasome is a large multi‑protein cluster that assembles inside immune cells when they detect danger. Its core consists of the sensor protein NLRP3, an adapter called ASC, and the enzyme caspase‑1. NLRP3 has several regions with distinct jobs: one end connects to ASC, the middle binds energy‑rich ATP and helps NLRP3 units cluster, and the tail helps keep the sensor off until trouble appears. When activated, NLRP3 molecules oligomerize—group together—and recruit ASC, which forms filament‑like scaffolds that then draw in caspase‑1. Once clustered, caspase‑1 switches on and cuts inactive precursors of the potent inflammatory messengers IL‑1β and IL‑18, as well as a protein called gasdermin D that can punch holes in the cell membrane, producing a fiery, often lethal inflammatory response called pyroptosis.
Two Green Lights: Getting Ready and Firing
Turning on NLRP3 is not a single event but a two‑step process. The first step, called priming, occurs when receptors that sense microbes or inflammatory hormones are engaged. These signals activate the transcription factor NF‑κB, which boosts production of NLRP3 itself and of the inactive cytokine precursors IL‑1β and IL‑18. At the same time, a web of chemical tweaks called post‑translational modifications—such as adding or trimming phosphate, ubiquitin, SUMO, acetyl, lipid, or ADP‑ribose groups—“licenses” NLRP3, placing it in a poised but not yet dangerous state. The second step, activation, is triggered by a wide array of stimuli: extracellular ATP, bacterial toxins, needle‑like crystals such as silica or uric acid, mitochondrial stress, lysosomal rupture, and more. These diverse triggers converge on common cellular changes—especially ion movements across the membrane, mitochondrial dysfunction, and damage to internal compartments—that drive the physical assembly of the inflammasome complex and full activation of caspase‑1.
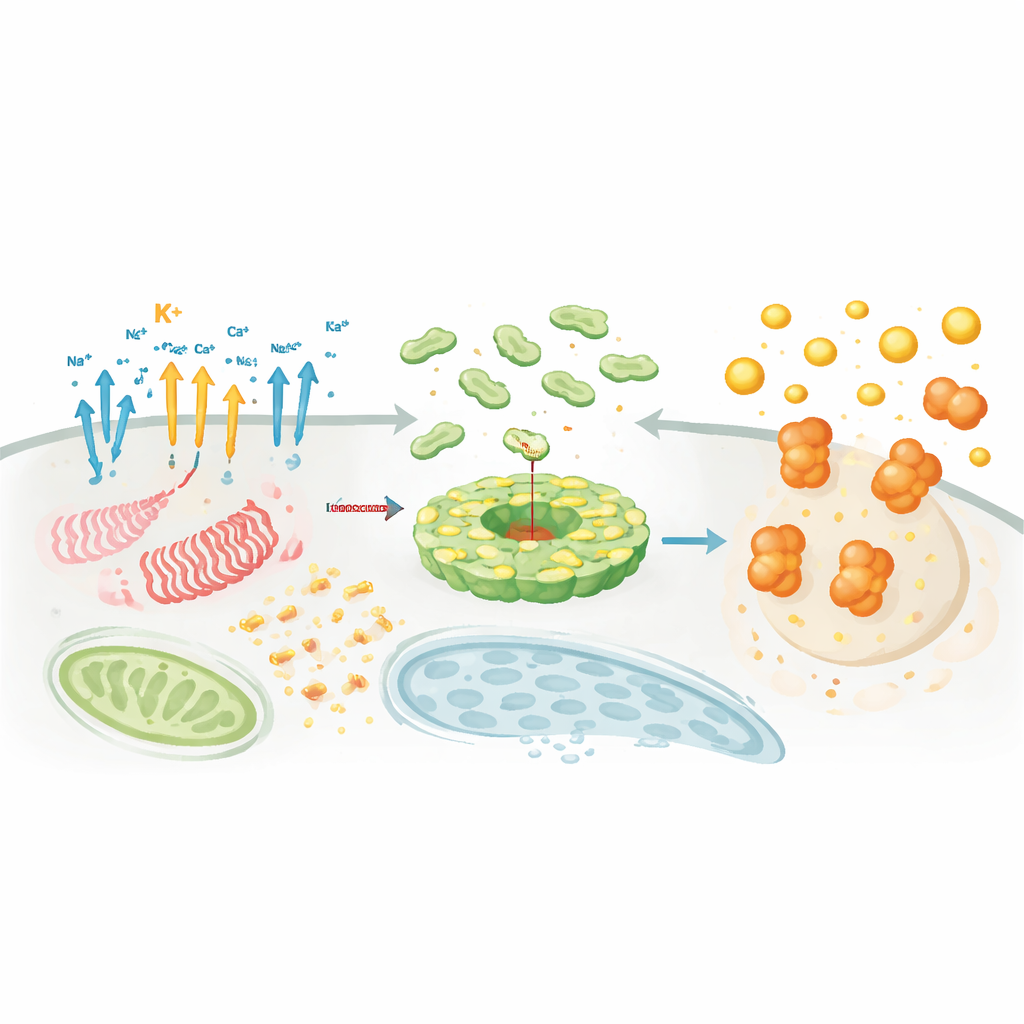
Ions, Powerhouses, and Recycling Centers Join the Story
Even though NLRP3 responds to many different threats, several core processes keep appearing. One is the flow of charged ions. Loss of potassium from the cell is the most consistent activator, helped or modulated by sodium, chloride, and calcium movements through specialized channels. Another is mitochondrial distress: damaged “powerhouses” release reactive oxygen species and fragments of mitochondrial DNA that help recruit and activate NLRP3, and drugs such as metformin can dampen this mitochondrial signal. A third is lysosomal injury, which can occur when cells engulf sharp particles; rupture of these recycling centers releases enzymes and ions that further promote NLRP3 activation. Meanwhile, changing cellular metabolism—how cells burn glucose, fats, and other fuels—can either accelerate or brake inflammasome activity, with some metabolites like succinate and palmitate driving inflammation and others like itaconate, fumarate, or ketone bodies restraining it.
Where and When the Complex Assembles
Beyond chemical signals, location and timing are crucial. NLRP3 and its partners move between organelles, hitchhiking along the cell’s internal scaffolding to gather at specific hubs such as the trans‑Golgi network and the microtubule‑organizing center. There, helper proteins like NEK7 act as “licensing factors” that reshape NLRP3 from an autoinhibited cage into an active disc that can bind ASC. Lipid modifications, like palmitoylation, and electrostatic interactions with particular membrane lipids steer NLRP3 to the right place at the right time. Parallel to this choreography, dozens of enzymes attach or remove regulatory tags—ubiquitin chains, phosphate groups, acetyl marks, and SUMO proteins—that fine‑tune NLRP3 abundance, stability, and its ability to hook up with ASC and caspase‑1. This dense regulatory network explains why NLRP3 can be exquisitely sensitive to context, cell type, and disease state.
Balancing Help and Harm
Because an overactive inflammasome can be as dangerous as an infection, cells also deploy brakes. Many proteins bind NLRP3 or its co‑factors to block key interactions, route it for degradation, or prevent its clustering, while others enhance its assembly under genuine threat. The review catalogues these positive and negative partners and shows that subtle defects—too much of an activating modification or too little of an inhibitory one—can tip the system toward chronic inflammation. The authors argue that understanding how all these switches interact will be essential for designing therapies. By targeting specific enzymes that modify NLRP3 or its helpers, future drugs could selectively cool the inflammasome in particular tissues or diseases, preserving its lifesaving role in host defense while preventing the collateral damage seen in many inflammatory and degenerative disorders.
Citation: Shin, H.J., Kim, I.S., Kim, J.K. et al. Molecular mechanisms of NLRP3 inflammasome activation. Exp Mol Med 58, 650–663 (2026). https://doi.org/10.1038/s12276-026-01656-9
Keywords: NLRP3 inflammasome, innate immunity, inflammation, post-translational modification, immunometabolism