Clear Sky Science · zh
STAT3 Thr714 磷酸化在 NPM-ALK 驱动的肿瘤形成中起关键作用
癌细胞内的微小开关如何改变其命运
癌症常常劫持机体的正常控制系统,将普通的细胞信号变成持续的生长和分裂指令。本研究聚焦于一种位于免疫细胞内的控制开关,这些免疫细胞参与一种称为间变性大细胞淋巴瘤(ALCL)的血液癌。通过放大观察附着在关键蛋白上的单一化学标记,研究者展示了看似微小的改变如何决定癌细胞是持续增殖还是被抑制。
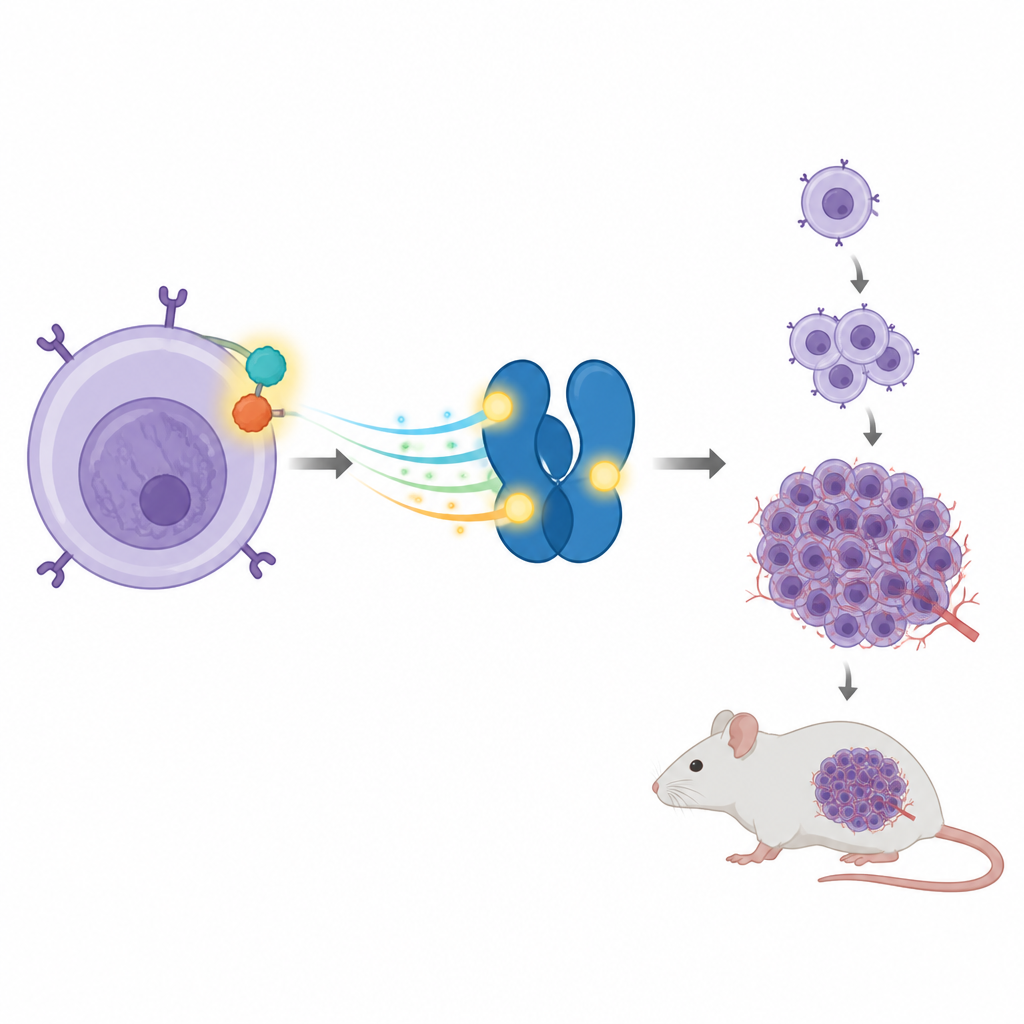
免疫细胞内推动癌变的合作关系
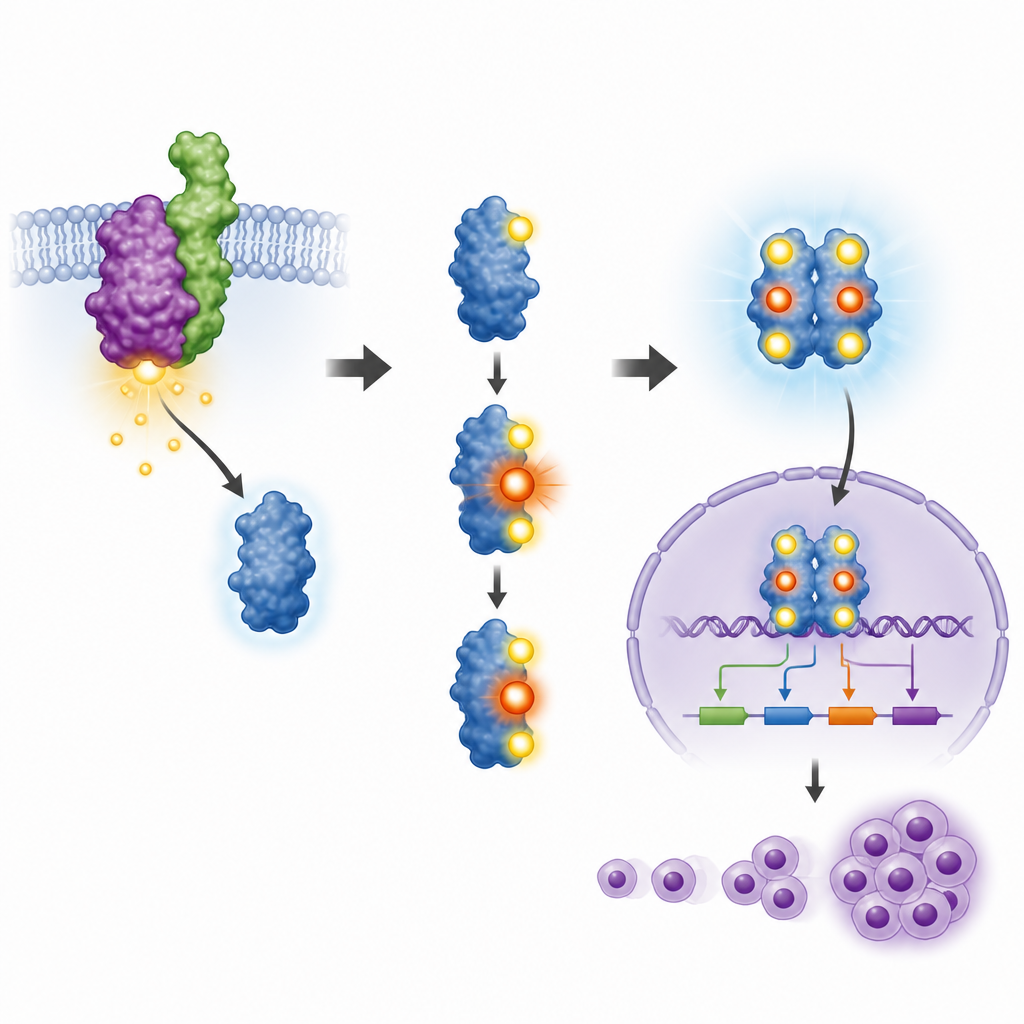
一些 ALCL 肿瘤携带断裂并重连的基因,融合成将两个蛋白拼接为一体的产物,称为 NPM-ALK。该杂合蛋白行为类似卡住的油门踏板,不断向细胞内部发送生长信号。它的一个主要靶点是另一种名为 STAT3 的蛋白,STAT3 通常帮助细胞响应外界信号,通过开启或关闭特定基因来调整行为。在 ALCL 中,NPM-ALK 持续激活 STAT3,进而增强那些推动细胞异常分裂和生存的基因表达。
熟悉蛋白上的新型控制位点
STAT3 仅在其某些微小位点被附加磷酸基(即磷酸化)时发挥作用。已知其中两个位点很重要,但第三个位点 T714 的作用尚不明确。研究团队制备了一种特异性抗体,仅识别在该位点被磷酸化的 STAT3,并用它来分析病人来源的癌细胞系和小鼠细胞模型。他们发现,当 NPM-ALK 活性存在时,STAT3 在包括 T714 在内的三个位点上均被磷酸化;用现有药物抑制 NPM-ALK 会去除这些磷酸化标记。这表明导致癌变的融合蛋白直接控制 T714,与已知的位点并列。
去掉 T714 开关会发生什么
为测试 T714 的实际功能,研究人员让小鼠血细胞表达 NPM-ALK,然后降低其内源性 STAT3 水平,并用正常型或在 T714 位置做了不能被磷酸化的突变体来替换。正常的 STAT3 在两个主要控制位点上被正确修饰,进入细胞核并激活与癌症相关的基因,如 Cyclin D1 和 Pim 家族基因。相比之下,T714 突变体仅获得了通常的其中一个磷酸化位点,大多停留在细胞质中,未能激活这些基因。因此,携带突变 STAT3 的细胞生长较慢、分裂减少,且未能得到 NPM-ALK 通常提供的那种对生长信号的独立性。
从细胞培养到活体小鼠的肿瘤形成
团队接着考察这一 STAT3 单个位点是否也影响活体动物中的肿瘤生长。他们将不同改造的细胞注入小鼠皮下。携带 NPM-ALK 和正常 STAT3 的细胞形成了大块肿瘤,并导致动物的肝脏和脾脏肿大,显示出侵袭性疾病。当敲低 STAT3 时,肿瘤生长显著下降。补回正常 STAT3 恢复了大部分致瘤能力,但补回 T714 突变体则未能恢复。接受携带突变蛋白细胞的小鼠,其肿瘤明显更小,器官大小接近正常。这些结果将 T714 处的微小磷酸化标记与该模型中的完整致癌能力联系了起来。
对未来癌症治疗的意义
总体而言,这些发现表明在 NPM-ALK 驱动的细胞中,T714 充当了 STAT3 的上游开启开关,帮助促成随后使 STAT3 进入细胞核并启动生长基因的改变。对非专业读者而言,这意味着除了针对癌症通路中显而易见的主要按钮外,也可以瞄准那些为激活做准备的隐性引发开关。设计阻断 STAT3 在 T714 位点被磷酸化的药物,或抑制执行该步骤的酶,可能为抑制 ALCL 中过度活跃的生长信号提供一种新策略,并可能适用于依赖类似融合蛋白的其他癌症。
引用: Lin, X., Yao, Y., Moriwaki, Y. et al. A critical role for STAT3 Thr714 phosphorylation in NPM-ALK-driven tumorigenesis. Sci Rep 16, 15005 (2026). https://doi.org/10.1038/s41598-026-44867-w
关键词: STAT3, NPM-ALK, 间变性大细胞淋巴瘤, 磷酸化, 肿瘤形成