Clear Sky Science · it
Un ruolo critico della fosforilazione STAT3 Thr714 nella tumorigenesi guidata da NPM-ALK
Come un piccolo interruttore nelle cellule tumorali può cambiare il loro destino
Il cancro spesso dirotta i normali sistemi di controllo dell’organismo, trasformando segnali cellulari ordinari in comandi continui di crescita e divisione. Questo studio esamina uno di questi interruttori all’interno delle cellule immunitarie coinvolte in un tipo di tumore del sangue chiamato linfoma anaplastico a grandi cellule (ALCL). Focalizzandosi su un singolo tag chimico applicato a una proteina chiave, i ricercatori mostrano come un cambiamento apparentemente piccolo possa decidere se le cellule tumorali continuano a moltiplicarsi o vengono tenute sotto controllo.
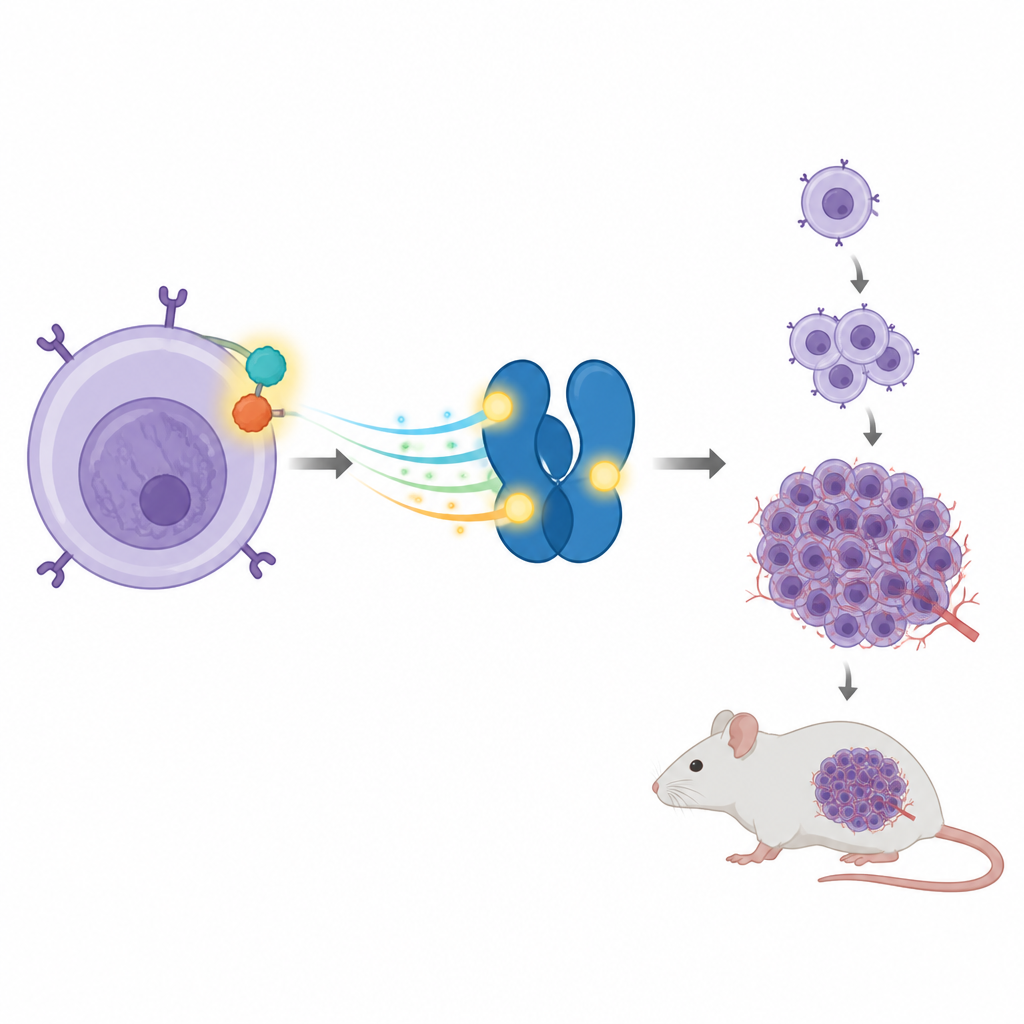
Una partnership che guida il cancro all’interno delle cellule immunitarie
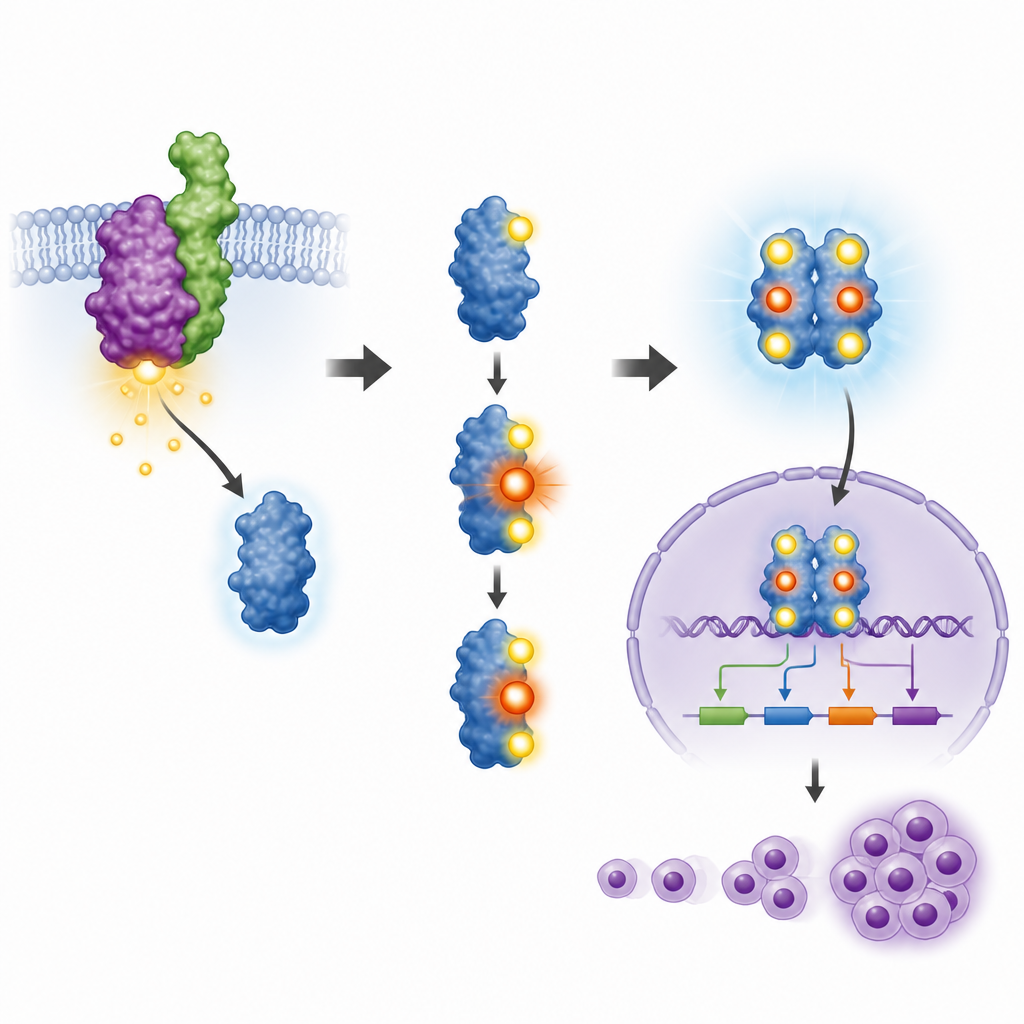
Alcuni tumori ALCL presentano un gene rotto e riunito che fonde due proteine in una, chiamata NPM‑ALK. Questa proteina ibrida si comporta come un acceleratore bloccato, inviando costantemente segnali di crescita all’interno della cellula. Uno dei suoi bersagli preferiti è un’altra proteina chiamata STAT3, che normalmente aiuta le cellule a rispondere a stimoli esterni accendendo o spegnendo geni specifici. Nell’ALCL, NPM‑ALK mantiene STAT3 costantemente attivato, il che a sua volta potenzia geni che spingono le cellule a dividersi e sopravvivere quando non dovrebbero.
Un nuovo punto di controllo su una proteina nota
STAT3 funziona solo quando su di essa sono presenti dei minuscoli siti marcati con gruppi fosfato, un processo chiamato fosforilazione. Due di questi siti erano già noti come importanti, ma il ruolo di un terzo sito, chiamato T714, era poco chiaro. Il gruppo ha creato un anticorpo su misura in grado di riconoscere STAT3 solo quando questo sito è marcato, e lo ha usato per studiare linee cellulari tumorali di pazienti e un modello cellulare murino. Hanno scoperto che quando NPM‑ALK era attivo, STAT3 risultava fosforilato in tutti e tre i siti, incluso T714, e che l’inibizione di NPM‑ALK con farmaci noti rimuoveva queste marcature. Ciò ha dimostrato che la proteina di fusione oncogena controlla direttamente T714 insieme ai siti più noti.
Cosa accade quando l’interruttore a T714 viene rimosso
Per verificare la funzione di T714, i ricercatori hanno ingegnerizzato cellule ematiche murine per esprimere NPM‑ALK, quindi hanno ridotto il livello di STAT3 endogeno e lo hanno sostituito con la versione normale o con una mutante in cui T714 era modificato in modo da non poter essere fosforilato. STAT3 normale veniva correttamente modificato nei due siti di controllo principali, si spostava nel nucleo e attivava geni associati al cancro come Cyclin D1 e geni della famiglia Pim. Al contrario, la variante mutante in T714 riceveva solo una delle consuete marcature, rimaneva per lo più fuori dal nucleo e non riusciva ad attivare questi geni. Di conseguenza, le cellule con STAT3 mutante crescevano più lentamente, si dividevano meno e non acquisivano la stessa indipendenza dai segnali di crescita che NPM‑ALK normalmente conferisce.
Dalla coltura cellulare ai tumori nei topi vivi
Il gruppo ha poi chiesto se questo singolo sito su STAT3 influenzasse anche la crescita tumorale in animali vivi. Hanno iniettato sotto la pelle dei topi le diverse cellule ingegnerizzate. Le cellule con NPM‑ALK e STAT3 normale formavano grandi tumori e causavano l’ingrossamento del fegato e della milza degli animali, riflettendo una malattia aggressiva. Quando STAT3 veniva silenziato, la crescita tumorale calava nettamente. Ripristinare STAT3 normale recuperava buona parte della capacità di formare tumori, mentre introdurre la variante T714 mutata non lo faceva. I topi riceventi cellule con la proteina mutante avevano tumori molto più piccoli e dimensioni degli organi quasi normali. Questi risultati collegano il piccolo gruppo fosfato su T714 al pieno potere oncogenico in questo modello.
Cosa significa per i futuri trattamenti contro il cancro
Complessivamente, i risultati suggeriscono che T714 agisca come un interruttore di avvio a monte per STAT3 nelle cellule guidate da NPM‑ALK, favorendo cambiamenti successivi che permettono a STAT3 di entrare nel nucleo e attivare geni della crescita. Per i non specialisti, ciò significa che, oltre a mirare solo ai pulsanti più visibili di una via oncogenica, potrebbe essere possibile puntare anche a un interruttore nascosto che prepara il sistema all’attivazione. Farmaci progettati per bloccare la marcatura di STAT3 in T714, o l’enzima che compie questo passaggio, potrebbero offrire un nuovo modo per silenziare un segnale di crescita sovraattivato nell’ALCL e, possibilmente, in altri tumori che dipendono da proteine di fusione simili.
Citazione: Lin, X., Yao, Y., Moriwaki, Y. et al. A critical role for STAT3 Thr714 phosphorylation in NPM-ALK-driven tumorigenesis. Sci Rep 16, 15005 (2026). https://doi.org/10.1038/s41598-026-44867-w
Parole chiave: STAT3, NPM-ALK, linfoma anaplastico a grandi cellule, fosforilazione, tumorigenesi