Clear Sky Science · en
A critical role for STAT3 Thr714 phosphorylation in NPM-ALK-driven tumorigenesis
How a tiny switch in cancer cells can change their fate
Cancer often hijacks the body’s normal control systems, turning ordinary cell signals into constant grow and divide commands. This study looks at one such control switch inside immune cells involved in a type of blood cancer called anaplastic large cell lymphoma (ALCL). By zooming in on a single chemical tag attached to a key protein, the researchers show how a seemingly small change can decide whether cancer cells keep multiplying or are held in check.
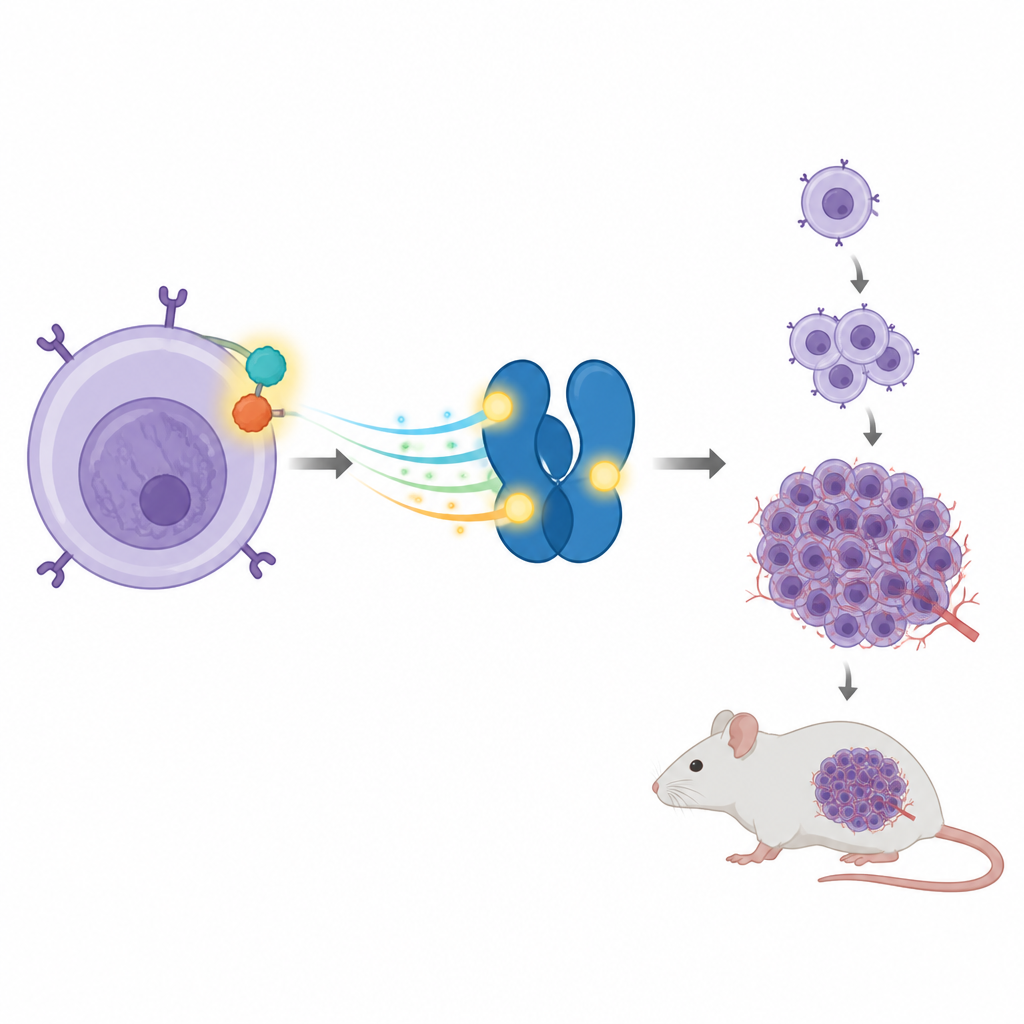
A cancer driving partnership inside immune cells
Some ALCL tumors carry a broken and rejoined gene that fuses two proteins into one, called NPM ALK. This hybrid protein behaves like a stuck accelerator pedal, constantly sending growth signals inside the cell. One of its favorite targets is another protein named STAT3, which normally helps cells respond to outside cues by turning specific genes on or off. In ALCL, NPM ALK keeps STAT3 switched on, which in turn boosts genes that push cells to divide and survive when they should not.
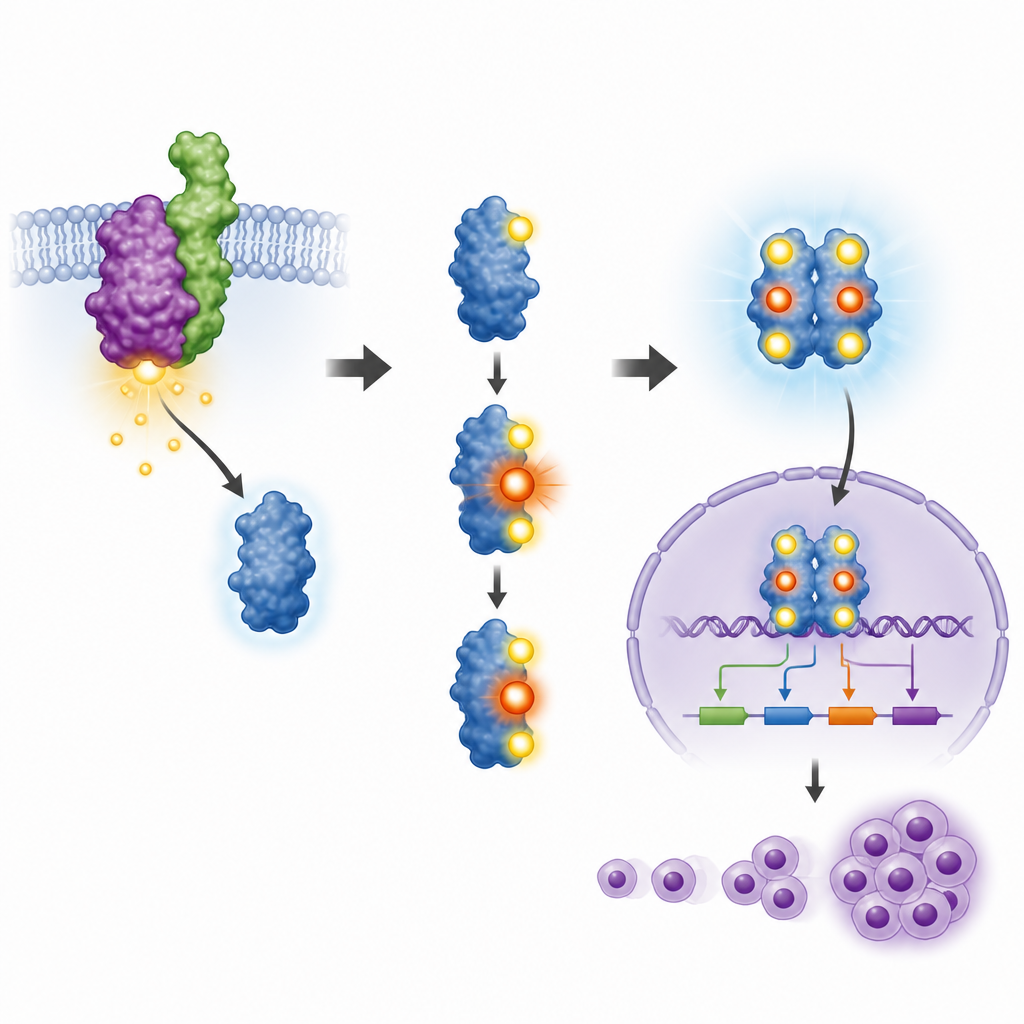
A new control spot on a familiar protein
STAT3 works only when certain tiny sites on it are tagged with phosphate groups, a process called phosphorylation. Two of these sites were already known to be important, but the role of a third site, called T714, was unclear. The team created a custom antibody that could recognize STAT3 only when this site was tagged, and used it to study cancer cell lines from patients and a mouse cell model. They found that when NPM ALK was active, STAT3 was tagged at all three sites, including T714, and that blocking NPM ALK with existing drugs removed these tags. This showed that the cancer driving fusion protein directly controls T714 alongside the better known sites.
What happens when the switch at T714 is removed
To test what T714 really does, the researchers engineered mouse blood cells to express NPM ALK, then reduced their natural STAT3 and replaced it either with the normal version or a mutant in which T714 was changed so it could not be tagged. Normal STAT3 became properly modified at its two main control sites, moved into the cell nucleus, and turned on cancer linked genes such as Cyclin D1 and Pim family genes. In contrast, the T714 mutant received only one of the usual tags, stayed mostly outside the nucleus, and failed to activate these genes. As a result, cells with mutant STAT3 grew more slowly, divided less often, and did not gain the same freedom from growth signals that NPM ALK usually provides.
From cell culture to tumors in living mice
The team then asked whether this single site on STAT3 also affects tumor growth in living animals. They injected the different engineered cells under the skin of mice. Cells with NPM ALK and normal STAT3 formed large tumors and caused the animals’ livers and spleens to swell, reflecting aggressive disease. When STAT3 was knocked down, tumor growth dropped sharply. Adding back normal STAT3 restored much of the tumor forming ability, but adding the T714 mutant did not. Mice given cells with the mutant protein had much smaller tumors and near normal organ sizes. These results linked the tiny phosphate tag at T714 to full cancer causing power in this model.
What this means for future cancer treatments
Together, the findings suggest that T714 acts as an upstream on switch for STAT3 in cells driven by NPM ALK, helping to enable later changes that let STAT3 enter the nucleus and switch on growth genes. For non specialists, this means that instead of targeting only the main visible buttons on a cancer pathway, it may also be possible to aim at a hidden primer switch that prepares the system for activation. Drugs designed to block the tagging of STAT3 at T714, or the enzyme that performs this step, could offer a new way to quiet an overactive growth signal in ALCL and possibly other cancers that rely on similar fusion proteins.
Citation: Lin, X., Yao, Y., Moriwaki, Y. et al. A critical role for STAT3 Thr714 phosphorylation in NPM-ALK-driven tumorigenesis. Sci Rep 16, 15005 (2026). https://doi.org/10.1038/s41598-026-44867-w
Keywords: STAT3, NPM-ALK, anaplastic large cell lymphoma, phosphorylation, tumorigenesis