Clear Sky Science · de
Eine entscheidende Rolle der STAT3-Thr714-Phosphorylierung bei der von NPM-ALK getriebenen Tumorentstehung
Wie ein winziger Schalter das Schicksal von Krebszellen verändern kann
Krebs kapert häufig die normalen Kontrollmechanismen des Körpers und verwandelt alltägliche Zellsignale in dauerhafte Aufforderungen zu Wachstum und Teilung. Diese Studie beschäftigt sich mit einem solchen Kontrollschalter in Immunzellen, die an einer Form von Blutkrebs beteiligt sind, dem anaplastischen großzelligen Lymphom (ALCL). Durch die genaue Untersuchung eines einzelnen chemischen Tags an einem Schlüsselprotein zeigen die Forscher, wie eine scheinbar kleine Veränderung darüber entscheiden kann, ob Krebszellen weiter proliferieren oder in Schach gehalten werden.
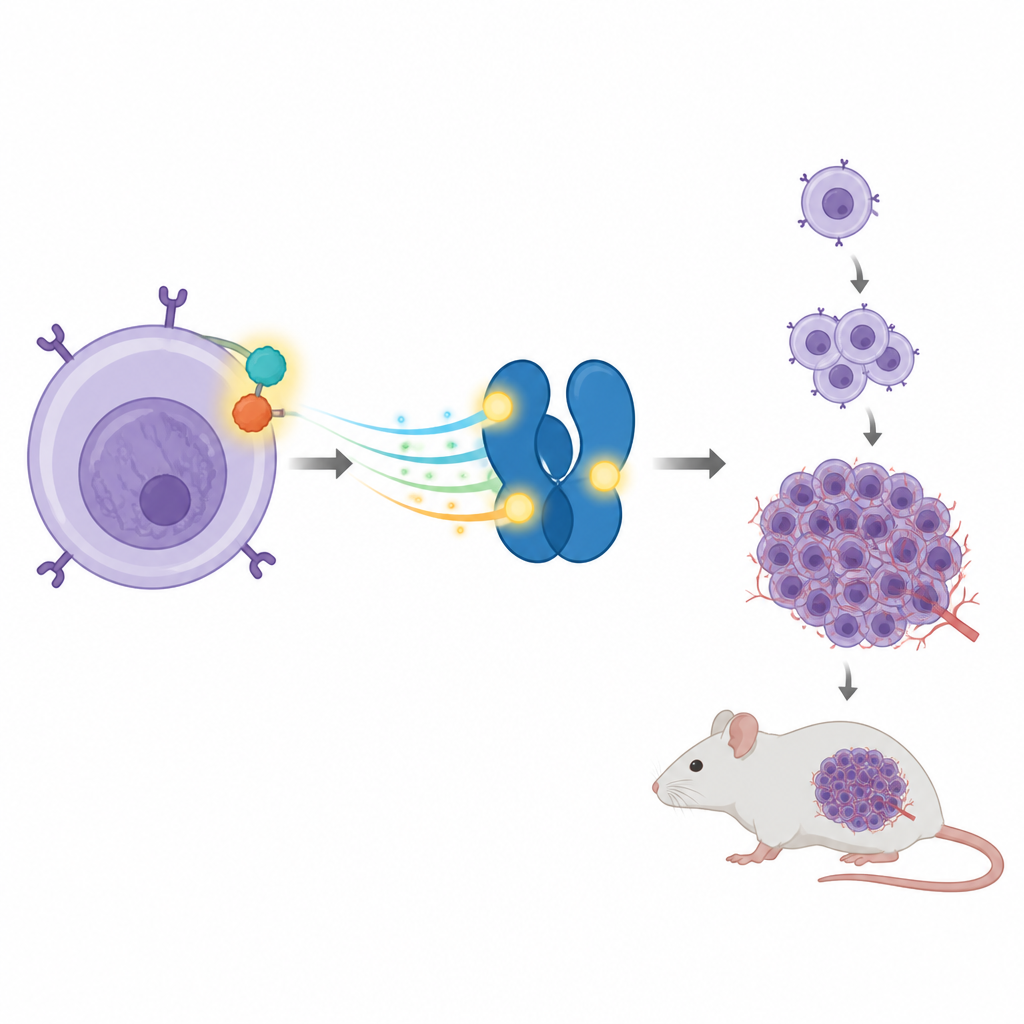
Eine treibende Partnerschaft in Immunzellen
Einige ALCL-Tumoren tragen ein gebrochenes und wieder zusammengefügtes Gen, das zwei Proteine zu einem verschmilzt, genannt NPM-ALK. Dieses Hybridprotein verhält sich wie ein festgestelltes Gaspedal und sendet ständig Wachstumssignale in die Zelle. Eines seiner bevorzugten Ziele ist ein anderes Protein namens STAT3, das normalerweise Zellen hilft, auf äußere Reize zu reagieren, indem es bestimmte Gene ein- oder ausschaltet. Bei ALCL hält NPM-ALK STAT3 dauerhaft aktiv, was wiederum Gene hochfährt, die die Zellen zur Teilung und zum Überleben befähigen, obwohl sie das nicht sollten.
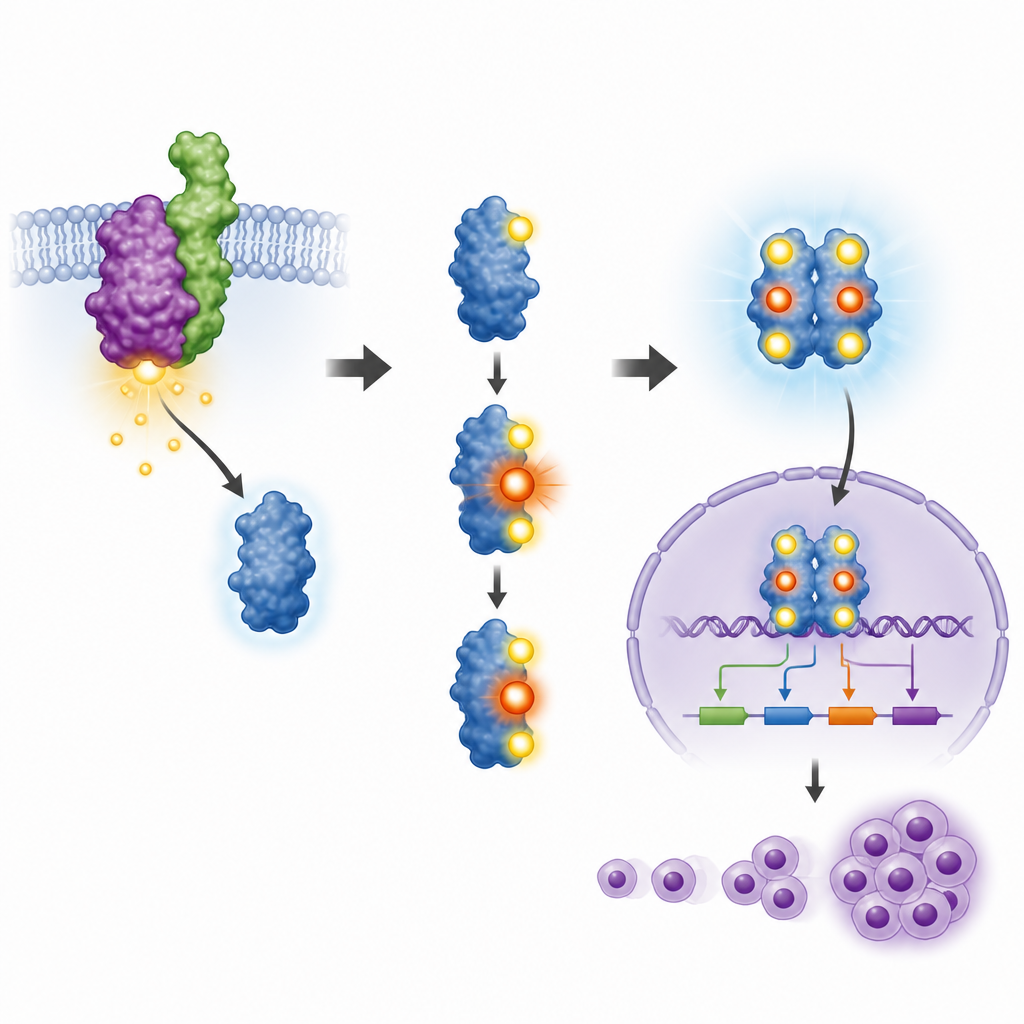
Eine neue Kontrollstelle an einem bekannten Protein
STAT3 ist nur dann funktionell, wenn bestimmte winzige Stellen daran mit Phosphatgruppen markiert sind, ein Prozess, der als Phosphorylierung bezeichnet wird. Zwei dieser Stellen waren bereits als wichtig bekannt, doch die Rolle einer dritten Stelle, genannt T714, war unklar. Das Team entwickelte einen maßgeschneiderten Antikörper, der STAT3 nur erkennt, wenn genau diese Stelle markiert ist, und setzte ihn an Zelllinien aus Patienten sowie an einem Mauszellmodell ein. Sie fanden heraus, dass bei aktiven NPM-ALK alle drei Stellen von STAT3 markiert waren, einschließlich T714, und dass die Blockade von NPM-ALK mit vorhandenen Wirkstoffen diese Markierungen entfernte. Das zeigte, dass das krebsantriebsende Fusionsprotein T714 direkt zusammen mit den bereits bekannten Stellen kontrolliert.
Was passiert, wenn der Schalter bei T714 entfernt wird
Um die Funktion von T714 zu prüfen, konstruierten die Forscher Mausblutzellen, die NPM-ALK exprimieren, reduzierten dann das endogene STAT3 und ersetzten es entweder durch die normale Version oder durch ein Mutant, bei dem T714 so verändert war, dass es nicht mehr markiert werden konnte. Das normale STAT3 wurde an seinen beiden Hauptkontrollstellen korrekt modifiziert, wanderte in den Zellkern und schaltete krebsassoziierte Gene wie Cyclin D1 und die Pim-Familie ein. Im Gegensatz dazu erhielt das T714-Mutant nur eine der üblichen Markierungen, verblieb größtenteils außerhalb des Zellkerns und aktivierte diese Gene nicht. Infolgedessen wuchsen Zellen mit dem mutierten STAT3 langsamer, teilten sich seltener und gewannen nicht die gleiche Unabhängigkeit von Wachstumssignalen, wie sie NPM-ALK normalerweise vermittelt.
Von Zellkultur zu Tumoren in lebenden Mäusen
Das Team prüfte anschließend, ob diese einzelne Stelle auf STAT3 auch das Tumorwachstum in lebenden Tieren beeinflusst. Sie injizierten die verschiedenen konstruierten Zellen unter die Haut von Mäusen. Zellen mit NPM-ALK und normalem STAT3 bildeten große Tumoren und verursachten eine Vergrößerung von Leber und Milz der Tiere, ein Zeichen aggressiver Erkrankung. Wurde STAT3 herunterreguliert, sank das Tumorwachstum deutlich. Das Wiedereinführen von normalem STAT3 stellte einen Großteil der Tumorbildefähigkeit wieder her, das T714-Mutant jedoch nicht. Mäuse, die Zellen mit dem mutierten Protein erhielten, entwickelten deutlich kleinere Tumoren und nahezu normale Organgrößen. Diese Ergebnisse verbanden das winzige Phosphat-Tag an T714 mit der vollen krebserzeugenden Wirkung in diesem Modell.
Was das für künftige Krebstherapien bedeutet
Zusammen deuten die Befunde darauf hin, dass T714 als ein vorlager Schalter für STAT3 in Zellen wirkt, die von NPM-ALK getrieben werden, und spätere Veränderungen ermöglicht, die STAT3 den Eintritt in den Zellkern und das Einschalten von Wachstumsgenen erlauben. Für Nichtfachleute bedeutet das, dass man statt nur die offensichtlichen Hauptknöpfe eines Krebswegs anzugreifen, auch einen verborgenen Primärschalter ins Visier nehmen könnte, der das System für die Aktivierung vorbereitet. Medikamente, die die Markierung von STAT3 an T714 oder das Enzym, das diesen Schritt ausführt, blockieren, könnten einen neuen Weg bieten, ein überaktives Wachstumssignal bei ALCL und möglicherweise anderen Krebserkrankungen mit ähnlichen Fusionsproteinen zu dämpfen.
Zitation: Lin, X., Yao, Y., Moriwaki, Y. et al. A critical role for STAT3 Thr714 phosphorylation in NPM-ALK-driven tumorigenesis. Sci Rep 16, 15005 (2026). https://doi.org/10.1038/s41598-026-44867-w
Schlüsselwörter: STAT3, NPM-ALK, anaplastisches großzelliges Lymphom, Phosphorylierung, Tumorentstehung