Clear Sky Science · fr
Un rôle critique de la phosphorylation de STAT3 Thr714 dans la tumorigénèse pilotée par NPM‑ALK
Comment un petit interrupteur dans les cellules cancéreuses peut changer leur destin
Le cancer détourne souvent les systèmes de contrôle normaux de l’organisme, transformant des signaux cellulaires ordinaires en ordres permanents de croissance et de division. Cette étude examine l’un de ces commutateurs de contrôle au sein des cellules immunitaires impliquées dans un type de cancer du sang appelé lymphome à grandes cellules anaplasiques (LGCA). En se penchant sur une seule marque chimique apposée sur une protéine clé, les chercheurs montrent comment un changement apparemment minime peut décider si les cellules cancéreuses continuent de se multiplier ou restent sous contrôle.
Un partenariat promoteur de cancer au sein des cellules immunitaires
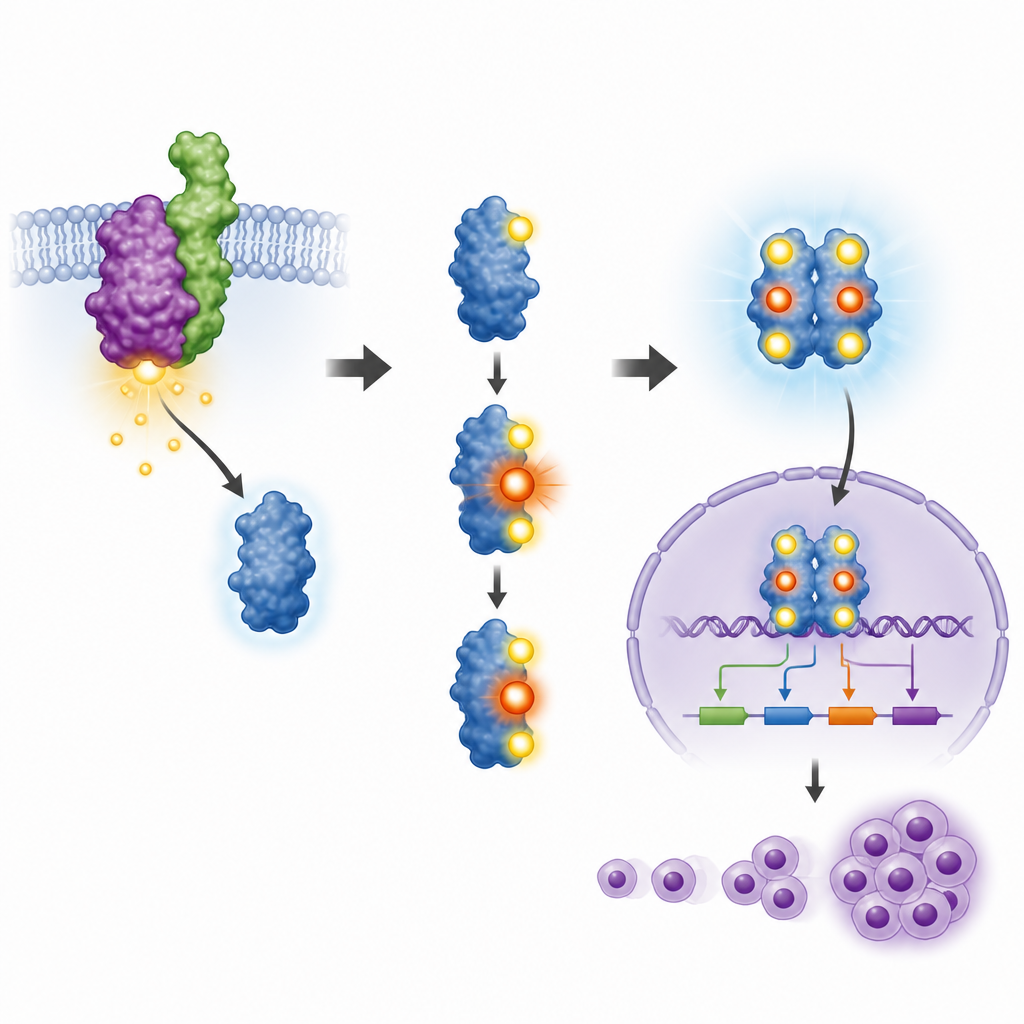
Certaines tumeurs de LGCA portent un gène cassé et réarrangé qui fusionne deux protéines en une seule, appelée NPM‑ALK. Cette protéine hybride fonctionne comme une pédale d’accélérateur bloquée, envoyant en permanence des signaux de croissance à l’intérieur de la cellule. L’une de ses cibles favorites est une autre protéine nommée STAT3, qui aide normalement les cellules à répondre à des signaux externes en allumant ou en éteignant des gènes spécifiques. Dans le LGCA, NPM‑ALK maintient STAT3 activé, ce qui à son tour renforce l’expression de gènes qui poussent les cellules à se diviser et à survivre alors qu’elles ne devraient pas.
Un nouveau point de contrôle sur une protéine bien connue
STAT3 n’est actif que lorsque certains sites minuscules y sont marqués par des groupes phosphate, un processus appelé phosphorylation. Deux de ces sites étaient déjà connus comme importants, mais le rôle d’un troisième site, appelé T714, restait flou. L’équipe a mis au point un anticorps spécifique capable de reconnaître STAT3 uniquement lorsque ce site est phosphorylé, et l’a utilisé pour étudier des lignées cellulaires de patients et un modèle de cellules de souris. Ils ont constaté que lorsque NPM‑ALK était actif, STAT3 était phosphorylé sur les trois sites, y compris T714, et que l’inhibition de NPM‑ALK avec des médicaments existants supprimait ces marquages. Cela montre que la protéine de fusion oncogénique contrôle directement T714 en parallèle des sites mieux connus.
Que se passe‑t‑il lorsque l’interrupteur T714 est retiré
Pour tester la fonction réelle de T714, les chercheurs ont modifié génétiquement des cellules sanguines de souris pour exprimer NPM‑ALK, puis ont réduit leur STAT3 naturel et l’ont remplacé soit par la version normale, soit par un mutant dans lequel T714 avait été modifié pour ne plus pouvoir être phosphorylé. STAT3 normal a été correctement phosphorylé sur ses deux principaux sites de contrôle, est entré dans le noyau cellulaire et a activé des gènes liés au cancer tels que Cyclin D1 et des gènes de la famille Pim. En revanche, le mutant T714 n’a reçu qu’un seul des marquages habituels, est resté majoritairement hors du noyau et n’a pas réussi à activer ces gènes. En conséquence, les cellules portant le STAT3 mutant ont crû plus lentement, se sont divisées moins souvent et n’ont pas acquis la même indépendance vis‑à‑vis des signaux de croissance que confère habituellement NPM‑ALK.
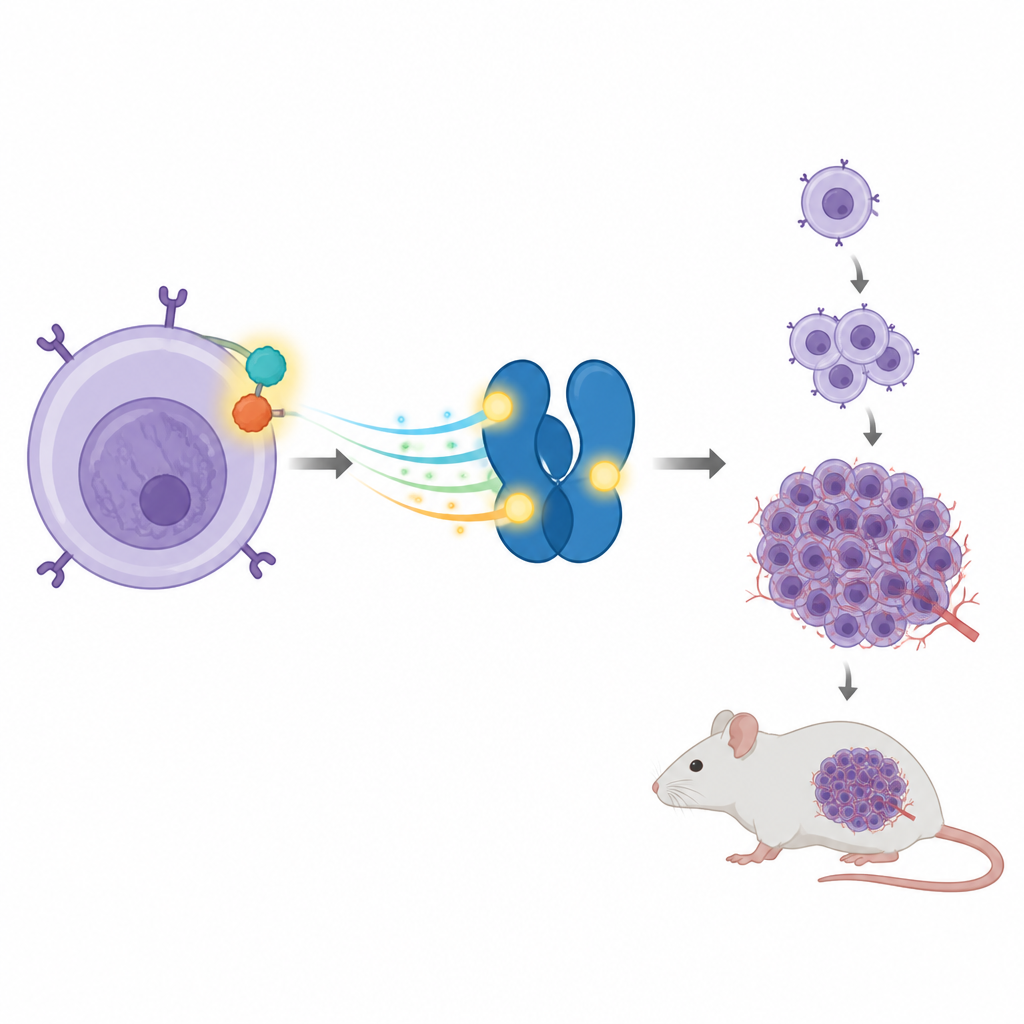
De la culture cellulaire aux tumeurs chez la souris
L’équipe a ensuite cherché à savoir si ce site unique sur STAT3 affectait aussi la croissance tumorale chez des animaux vivants. Ils ont injecté sous la peau des souris les différentes cellules modifiées. Les cellules exprimant NPM‑ALK et STAT3 normal ont formé de grosses tumeurs et provoqué une augmentation du volume du foie et de la rate des animaux, signes d’une maladie agressive. Lorsque STAT3 a été réduit, la croissance tumorale a fortement diminué. La réintroduction de STAT3 normal a rétabli une grande partie de la capacité tumorale, mais la réintroduction du mutant T714 ne l’a pas fait. Les souris recevant des cellules portant la protéine mutante avaient des tumeurs beaucoup plus petites et des organes proches de la taille normale. Ces résultats relient la petite marque phosphate en T714 à la pleine puissance oncogénique dans ce modèle.
Ce que cela signifie pour les traitements anticancéreux futurs
Ensemble, ces résultats suggèrent que T714 agit comme un interrupteur précoce activant STAT3 dans les cellules pilotées par NPM‑ALK, facilitant des changements ultérieurs qui permettent à STAT3 d’entrer dans le noyau et d’activer des gènes de croissance. Pour le grand public, cela signifie que, plutôt que de viser uniquement les boutons principaux visibles d’une voie oncogénique, il pourrait aussi être possible de cibler un interrupteur d’amorçage caché qui prépare le système à l’activation. Des médicaments conçus pour bloquer la phosphorylation de STAT3 en T714, ou l’enzyme qui réalise cette modification, pourraient offrir une nouvelle manière d’atténuer un signal de croissance suractivé dans le LGCA et potentiellement dans d’autres cancers dépendant de protéines de fusion similaires.
Citation: Lin, X., Yao, Y., Moriwaki, Y. et al. A critical role for STAT3 Thr714 phosphorylation in NPM-ALK-driven tumorigenesis. Sci Rep 16, 15005 (2026). https://doi.org/10.1038/s41598-026-44867-w
Mots-clés: STAT3, NPM-ALK, lymphome à grandes cellules anaplasiques, phosphorylation, tumorigénèse