Clear Sky Science · sv
En kritisk roll för STAT3 Thr714-fosforylering i NPM-ALK-driven tumörbildning
Hur en liten strömbrytare i cancerceller kan ändra deras öde
Cancer kapar ofta kroppens normala kontrollsystem och förvandlar vanliga cellulära signaler till ständiga kommandon om att växa och dela sig. Denna studie granskar en sådan kontrollpunkt i immunceller som är involverade i en typ av blodcancer kallad anaplastiskt storcelligt lymfom (ALCL). Genom att zooma in på en enda kemisk märkning på ett nyckelprotein visar forskarna hur en till synes liten förändring kan avgöra om cancerceller fortsätter att multiplicera sig eller hålls i schack.
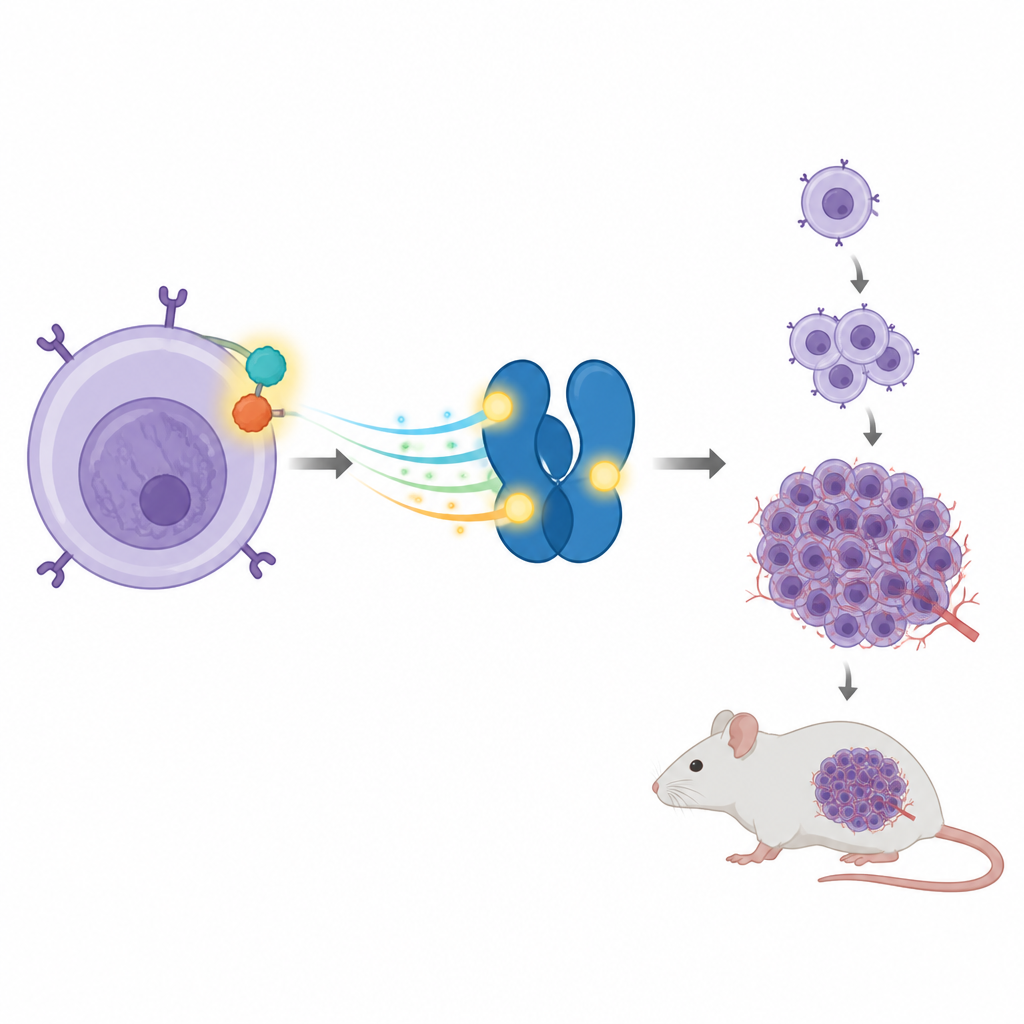
En cancerdrivande samverkan i immunceller
Vissa ALCL-tumörer bär på en brutet och återförenad gen som fuserar två proteiner till ett, kallat NPM-ALK. Detta hybridprotein beter sig som ett fastnat gaspedal och skickar konstant tillväxtsignaler inuti cellen. En av dess favoritmål är ett annat protein som heter STAT3, som normalt hjälper celler att svara på yttre signaler genom att slå på eller av specifika gener. I ALCL håller NPM-ALK STAT3 påkopplat, vilket i sin tur förstärker gener som driver celler att dela sig och överleva när de egentligen inte borde.
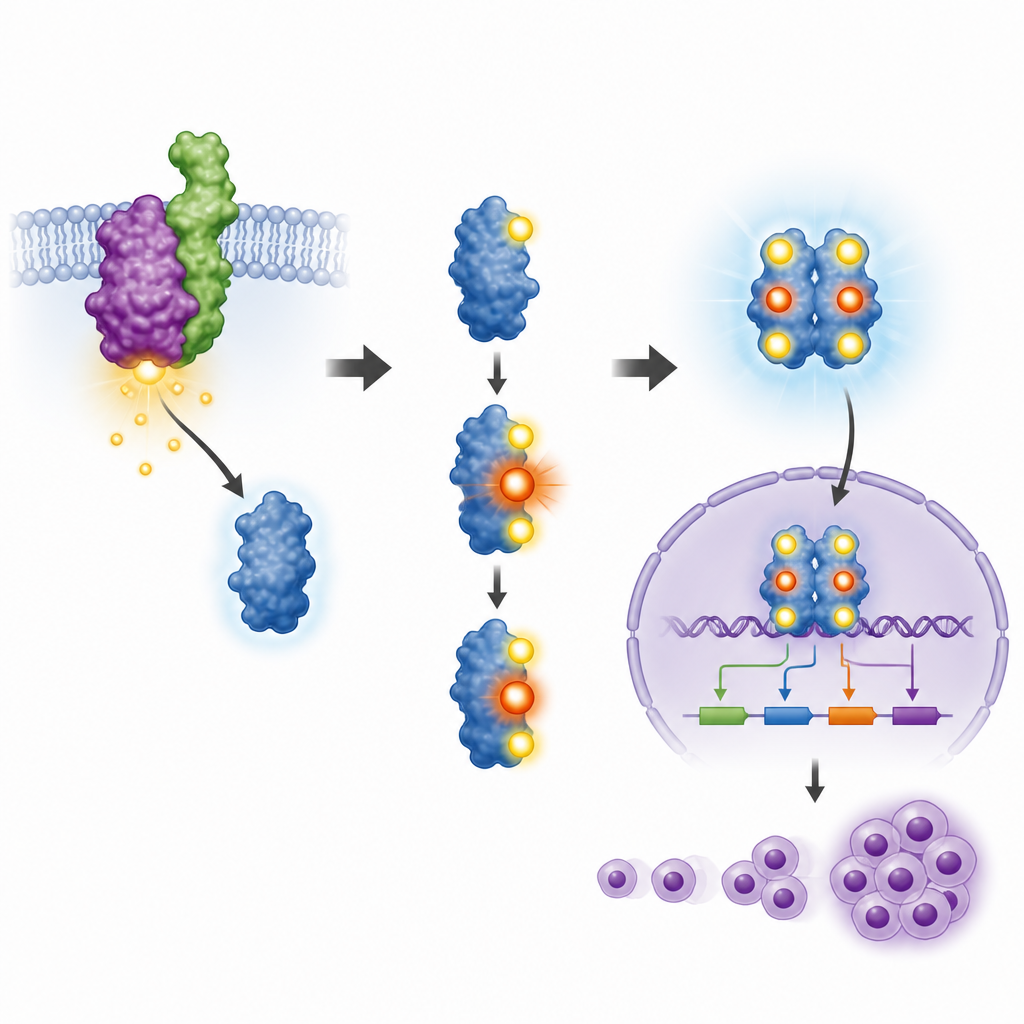
En ny kontrollpunkt på ett välkänt protein
STAT3 fungerar endast när vissa små platser på det är märkta med fosfatgrupper, en process som kallas fosforylering. Två av dessa platser var redan kända för att vara viktiga, men rollen för en tredje plats, kallad T714, var oklar. Forskarna skapade ett specialanpassat antikropp som kunde känna igen STAT3 endast när denna plats var märkt, och använde den för att studera cancercellinjer från patienter och en muscellmodell. De fann att när NPM-ALK var aktivt var STAT3 märkt vid alla tre platserna, inklusive T714, och att blockering av NPM-ALK med befintliga läkemedel avlägsnade dessa märkningar. Detta visade att det cancerdrivande fusionsproteinet direkt kontrollerar T714 tillsammans med de mer välkända platserna.
Vad som händer när strömbrytaren vid T714 tas bort
För att testa vad T714 verkligen gör konstruerade forskarna musblodceller för att uttrycka NPM-ALK, sänkte sedan deras naturliga STAT3 och ersatte det antingen med den normala varianten eller en mutant där T714 ändrats så att det inte kunde märkas. Normal STAT3 blev korrekt modifierad vid sina två huvudkontrollplatser, förflyttades till cellkärnan och aktiverade cancerrelaterade gener såsom Cyclin D1 och Pim-familjens gener. I kontrast fick T714-mutanten endast en av de vanliga märkningarna, stannade mestadels utanför kärnan och misslyckades med att aktivera dessa gener. Som ett resultat växte celler med mutant STAT3 långsammare, delade sig mer sällan och uppnådde inte samma frihet från tillväxtbegränsningar som NPM-ALK vanligtvis ger.
Från cellodling till tumörer i levande möss
Forskarna frågade sedan om denna enda plats på STAT3 också påverkar tumörtillväxt i levande djur. De injicerade de olika konstruerade cellerna under huden på möss. Celler med NPM-ALK och normal STAT3 bildade stora tumörer och fick djurens lever och mjälte att svullna, vilket speglar aggressiv sjukdom. När STAT3 slogs ned minskade tumörtillväxten kraftigt. Återinförande av normal STAT3 återställde mycket av tumörbildningsförmågan, men tillsats av T714-mutanten gjorde det inte. Möss som fick celler med det mutanta proteinet hade mycket mindre tumörer och nära normala organstorlekar. Dessa resultat kopplade den lilla fosfatmarkeringen vid T714 till full cancerframkallande kraft i denna modell.
Vad detta betyder för framtida cancerbehandlingar
Tillsammans tyder fynden på att T714 fungerar som en upstream på-knapp för STAT3 i celler drivna av NPM-ALK, vilket hjälper till att möjliggöra senare förändringar som låter STAT3 gå in i kärnan och slå på tillväxtgener. För icke-specialister innebär detta att istället för att bara rikta in sig på de huvudsakliga synliga knapparna i en cancerväg kan man också sikta på en dold primer-strömbrytare som förbereder systemet för aktivering. Läkemedel utformade för att blockera märkningen av STAT3 vid T714, eller enzymet som utför detta steg, skulle kunna erbjuda ett nytt sätt att dämpa en överaktiv tillväxtsignal i ALCL och möjligen andra cancerformer som förlitar sig på liknande fusionsproteiner.
Citering: Lin, X., Yao, Y., Moriwaki, Y. et al. A critical role for STAT3 Thr714 phosphorylation in NPM-ALK-driven tumorigenesis. Sci Rep 16, 15005 (2026). https://doi.org/10.1038/s41598-026-44867-w
Nyckelord: STAT3, NPM-ALK, anaplastiskt storcelligt lymfom, fosforylering, tumörbildning