Clear Sky Science · zh
ITPR3通过Ca2+/NF-κB/LECT2通路损伤肝细胞从而促进肝纤维化
为何肝脏瘢痕化重要
全球范围内肝病发病率上升,常常在多年无明显症状的情况下悄然进展。一个关键转折点是肝纤维化——瘢痕组织的堆积,最终可能发展为肝硬化和肝衰竭。本研究提出了一个关键问题:究竟是什么驱动了最早期的肝细胞损伤,从而启动这一瘢痕形成过程?阻断一个单一的分子“开关”能否帮助保护肝脏免受长期损害?
深入了解肝脏受损的机制
纤维化始于肝脏主要功能细胞——肝细胞的反复损伤。在这项研究中,研究人员使用一种成熟的化学物质四氯化碳,在小鼠及培养的鼠肝细胞中模拟慢性肝损伤。处理后的小鼠出现了与人类肝病相似的特征:血液中肝损伤标志物升高、脂质和胆固醇紊乱、炎症以及大量类瘢痕物质的沉积,破坏了正常的肝脏结构。这些改变证实了该模型能够忠实重现从单次损伤到持久瘢痕化的病理链条。
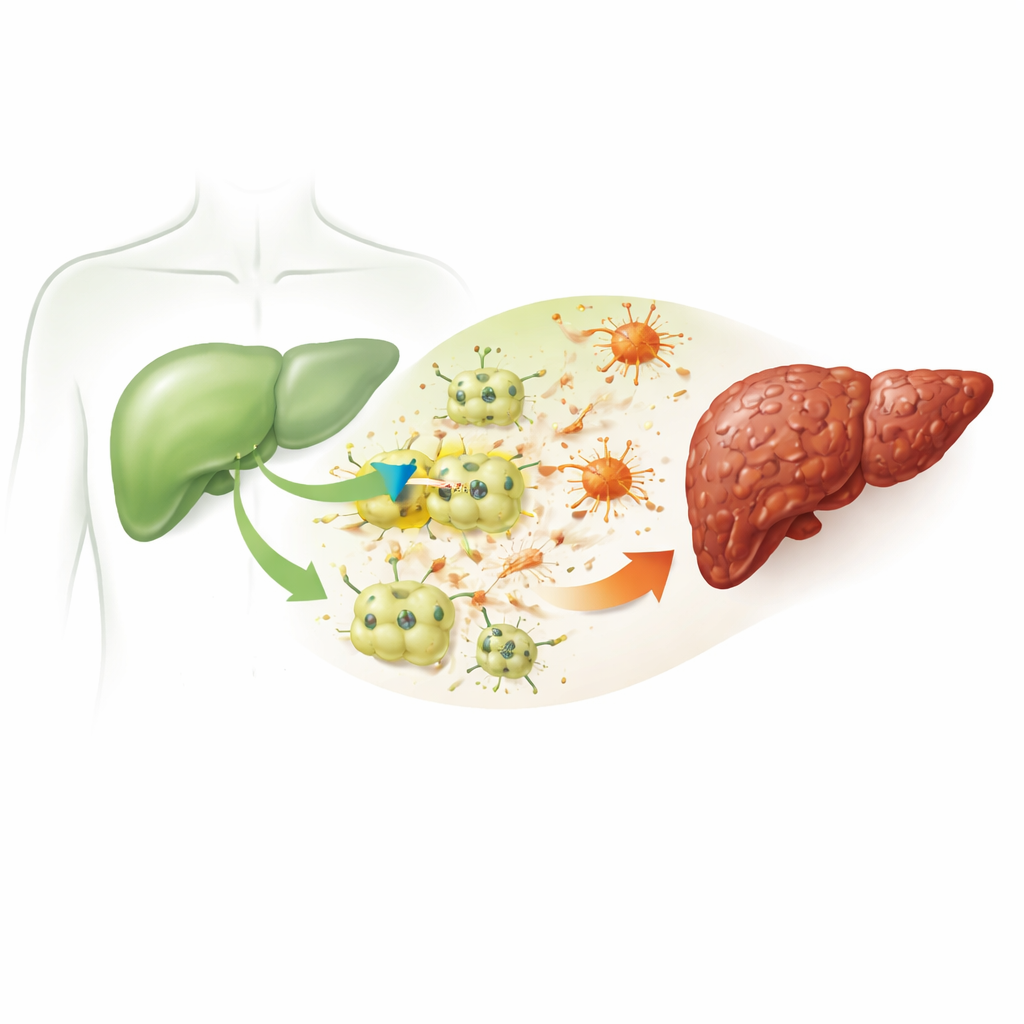
病变肝细胞中出现的隐秘通道
团队关注于一种名为ITPR3的蛋白——一种将钙从细胞内储存库释放出来的通道。健康肝细胞通常不表达这种通道。然而,在受损的小鼠肝脏和体外化学应激的培养肝细胞中,研究者发现ITPR3水平显著上升。在ITPR3出现的部位,肝细胞更容易发生程序性细胞死亡(即受控的自我毁灭)。当科学家使用基因干预工具(siRNA)下调ITPR3时,细胞内钙的激增消失,细胞死亡率显著下降。来自这些受损肝细胞的培养上清液还可以将邻近的支持细胞——肝星状细胞,诱导为活跃的、产生瘢痕的状态;而当抑制ITPR3时,这一激活效应被减弱。
从钙信号到炎症开关
钙不仅是结构性矿物质;在细胞内,短暂的钙脉冲作为信号开启或关闭通路。作者证明,通过新出现的ITPR3通道释放的过量钙会激活一个著名的炎症调控因子NF-κB。在受损小鼠肝脏和培养肝细胞中,NF-κB从细胞质转移到细胞核——这是其活化的标志。降低ITPR3水平可阻止这种核内转位,而化学抑制NF-κB则能减少细胞死亡。这些实验将ITPR3的出现与钙激增、NF-κB激活以及最终的肝细胞丧失联系起来,为瘢痕形成奠定了基础。
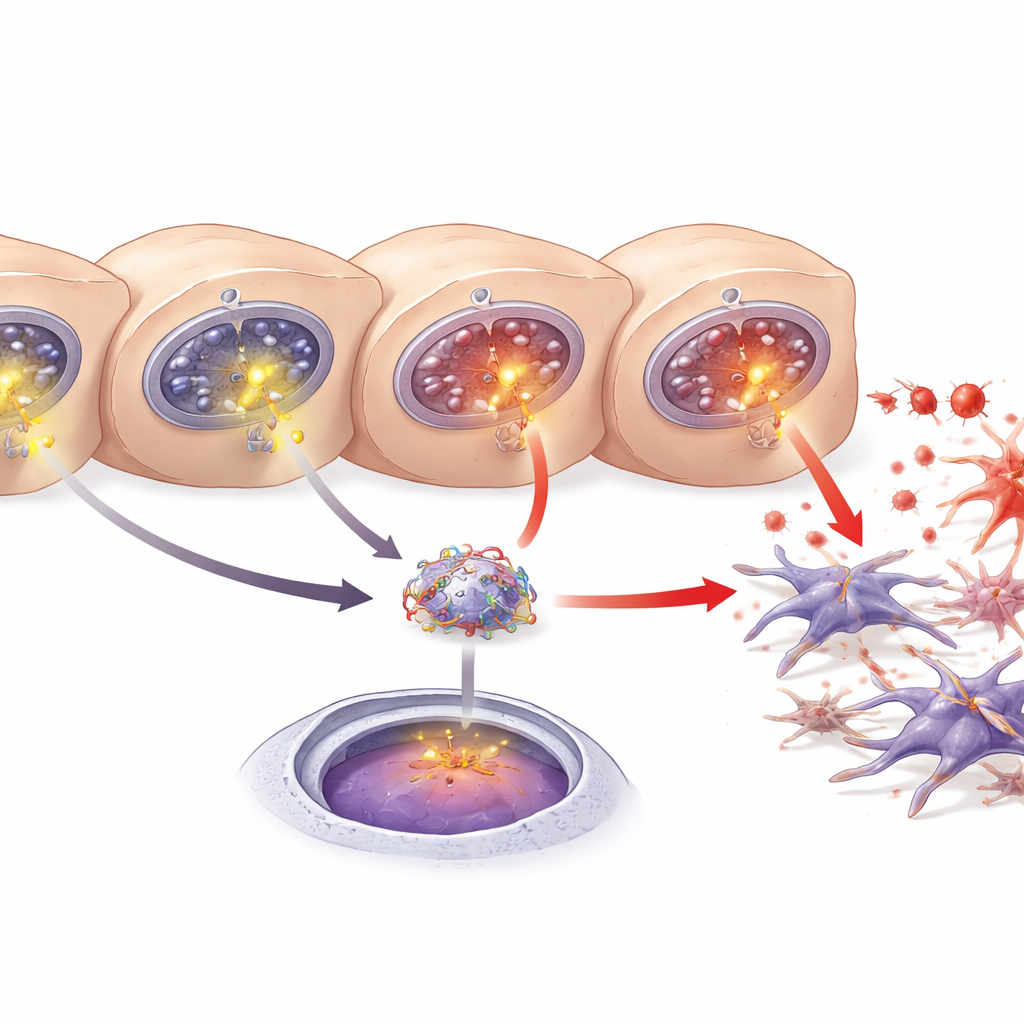
助长瘢痕化的肝脏信使
另一个关键参与者是来源于肝脏的蛋白LECT2,先前已被牵涉到脂肪肝、纤维化和肝癌。研究人员发现,当肝细胞受损时,血液、肝组织和培养基中的LECT2含量增加。通过数据库预测和遗传报告基因实验,他们表明NF-κB可以结合到LECT2基因调控区的一段特定位点,从而增强其活性。抑制NF-κB或下调ITPR3均能减少LECT2的产生和释放。综合来看,研究描绘出一条链条:在应激肝细胞中出现ITPR3,触发额外的钙释放,激活NF-κB,促使LECT2产生,进而促进星状细胞活化和纤维化。
对未来治疗的意义
简而言之,该研究将异常的钙通道ITPR3识别为一种上游的“开启开关”,它将持续的肝损伤转化为持久的瘢痕。通过展示ITPR3通过特定的钙–NF-κB–LECT2通路导致细胞死亡和纤维化,这项工作突出了若干潜在的药物靶点:通道本身、炎症开关或下游的肝信使。尽管在人体中还需更多研究来确认安全性和实际疗效,但这些结果表明关闭ITPR3可能有助于保护肝细胞、抑制炎症并减缓或阻止慢性肝病向肝硬化的进展。
引用: Zhao, X., Liu, X., Ni, M. et al. ITPR3 promotes liver fibrosis by damaging hepatocytes via the Ca2+/NF-B/LECT2 pathway. Sci Rep 16, 13376 (2026). https://doi.org/10.1038/s41598-026-43866-1
关键词: 肝纤维化, 肝细胞凋亡, 钙信号, NF-kappaB通路, LECT2