Clear Sky Science · it
ITPR3 promuove la fibrosi epatica danneggiando gli epatociti tramite la via Ca2+/NF-κB/LECT2
Perché la cicatrizzazione del fegato conta
Le malattie del fegato sono in aumento a livello globale e spesso progrediscono silenziosamente per anni prima che compaiano i sintomi. Un punto di svolta cruciale è la fibrosi epatica, l’accumulo di tessuto cicatriziale che può infine portare a cirrosi e insufficienza epatica. Questo studio pone una domanda fondamentale: che cosa provoca esattamente il danno iniziale agli epatociti che avvia questo processo di cicatrizzazione, e potrebbe il blocco di un singolo “interruttore” molecolare proteggere il fegato da danni a lungo termine?
Uno sguardo più ravvicinato a come il fegato viene danneggiato
La fibrosi ha inizio quando le principali cellule funzionali del fegato, chiamate epatociti, subiscono ripetuti danni. In questo lavoro i ricercatori hanno usato un noto agente chimico, il tetracloruro di carbonio, per riprodurre il danno epatico cronico in topi e in cellule epatiche murine coltivate. I topi trattati hanno sviluppato i segni caratteristici di malattia epatica osservati nell’uomo: marcatori ematici elevati di danno epatico, alterazioni di grassi e colesterolo, infiammazione e ingenti depositi di materiale cicatriziale che deformano l’architettura normale del fegato. Questi cambiamenti confermano che il modello riproduce fedelmente la catena di eventi che trasforma un danno semplice in una cicatrizzazione persistente.
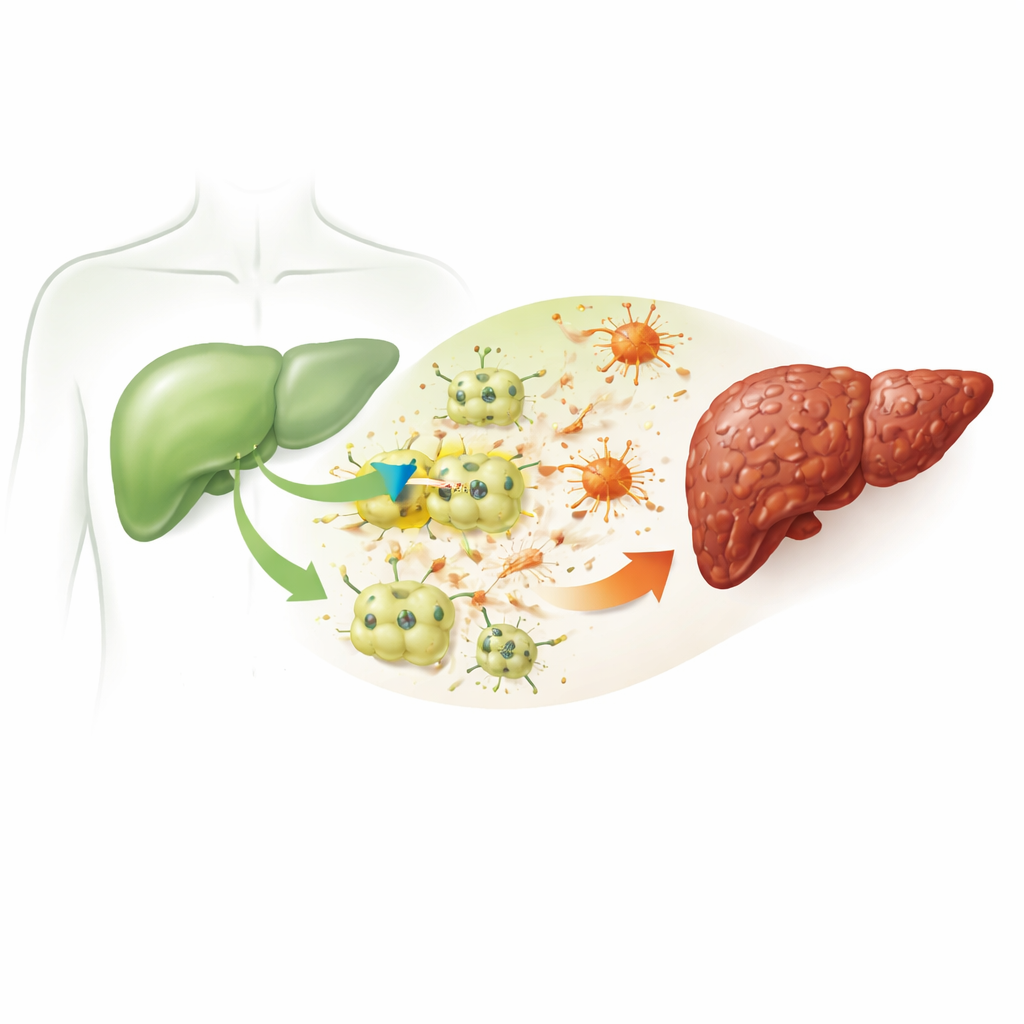
Un canale nascosto che compare nelle cellule epatiche malate
Il gruppo si è concentrato su una proteina chiamata ITPR3, un canale che rilascia calcio da depositi intracellulari. Gli epatociti sani normalmente non esprimono questo canale. Tuttavia, sia nei fegati danneggiati dei topi sia nelle cellule epatiche stressate chimicamente in coltura, i ricercatori hanno riscontrato un netto aumento dei livelli di ITPR3. Là dove compariva ITPR3, le cellule epatiche mostravano una maggiore probabilità di andare incontro a morte cellulare programmata, una forma controllata di autodistruzione. Quando gli scienziati hanno utilizzato uno strumento genetico (siRNA) per ridurre l’ITPR3, l’ondata di calcio intracellulare è scomparsa e il tasso di morte cellulare è diminuito nettamente. Il fluido rilasciato da questi epatociti danneggiati poteva inoltre trasformare le cellule di supporto vicine, le cellule stellate epatiche, in uno stato attivo produttrice di cicatrici — un cambiamento attenuato quando ITPR3 veniva soppresso.
Dal segnale del calcio all’interruttore infiammatorio
Il calcio non è solo un minerale strutturale; all’interno delle cellule i brevi impulsi di calcio fungono da segnali che attivano o disattivano vie cellulari. Gli autori hanno mostrato che l’eccesso di calcio rilasciato attraverso i canali ITPR3 neoformati attiva un noto regolatore dell’infiammazione, NF-κB. Nei fegati danneggiati dei topi e negli epatociti coltivati, NF-κB si è spostato dal citoplasma al nucleo, dove vengono controllati i geni, un marcatore della sua attivazione. Ridurre i livelli di ITPR3 ha impedito questo spostamento nucleare, mentre un inibitore chimico di NF-κB ha ridotto la morte cellulare. Questi esperimenti collegano la comparsa di ITPR3 a un’ondata di calcio, all’attivazione di NF-κB e, in ultima istanza, alla perdita di epatociti, predisponendo alla formazione di tessuto cicatriziale.
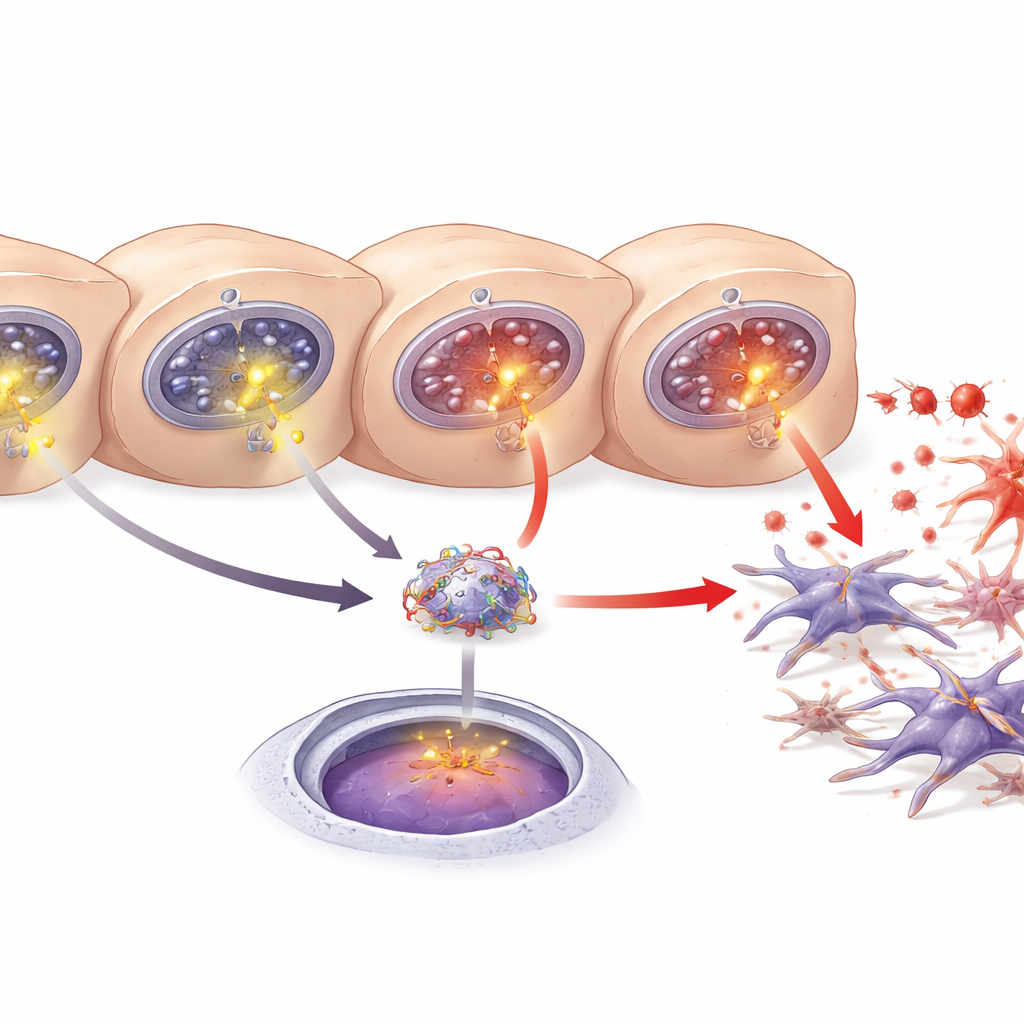
Un messaggero epatico che alimenta la cicatrizzazione
Un altro attore chiave è una proteina di origine epatica chiamata LECT2, precedentemente implicata nella steatosi epatica, nella fibrosi e nel cancro del fegato. I ricercatori hanno trovato livelli aumentati di LECT2 nel sangue, nel tessuto epatico e nel mezzo di coltura quando gli epatociti erano danneggiati. Utilizzando predizioni da database e test con reporter genetici, hanno dimostrato che NF-κB può legarsi a una specifica sequenza di DNA nella regione regolatrice del gene LECT2, potenziandone l’attività. Bloccare NF-κB o abbassare ITPR3 riduceva sia la produzione sia il rilascio di LECT2. Nel complesso, i risultati delineano una catena causale: ITPR3 compare negli epatociti stressati, provoca un rilascio di calcio aggiuntivo, attiva NF-κB, stimola la produzione di LECT2 e promuove l’attivazione delle cellule stellate e la fibrosi.
Che cosa significa per i trattamenti futuri
In termini semplici, lo studio identifica un canale del calcio anomalo, ITPR3, come un “interruttore” a monte che trasforma il danno epatico continuo in cicatrizzazione persistente. Mostrando che ITPR3 conduce a morte cellulare e fibrosi attraverso una specifica via calcio–NF-κB–LECT2, il lavoro evidenzia diversi possibili bersagli farmacologici: il canale stesso, l’interruttore infiammatorio o il messaggero epatico a valle. Sebbene siano necessarie più ricerche nell’uomo, soprattutto per confermare sicurezza e benefici nella pratica clinica, i risultati suggeriscono che spegnere ITPR3 potrebbe aiutare a preservare gli epatociti, calmare l’infiammazione e rallentare o prevenire la progressione della malattia epatica cronica verso la cirrosi.
Citazione: Zhao, X., Liu, X., Ni, M. et al. ITPR3 promotes liver fibrosis by damaging hepatocytes via the Ca2+/NF-B/LECT2 pathway. Sci Rep 16, 13376 (2026). https://doi.org/10.1038/s41598-026-43866-1
Parole chiave: fibrosi epatica, apoptosi degli epatociti, segnalazione del calcio, via NF-kappaB, LECT2