Clear Sky Science · de
ITPR3 fördert Leberfibrose, indem es Hepatozyten über den Ca2+/NF-κB/LECT2-Weg schädigt
Warum Lebervernarbung wichtig ist
Lebererkrankungen nehmen weltweit zu und verlaufen oft jahrelang still, bevor Symptome auftreten. Ein entscheidender Wendepunkt ist die Leberfibrose, die Ansammlung von Narbengewebe, die schließlich zur Zirrhose und zum Leberversagen führen kann. Diese Studie stellt eine zentrale Frage: Was genau verursacht die frühesten Schäden an Leberzellen, die diesen Vernarbungsprozess in Gang setzen, und könnte das Blockieren eines einzigen molekularen „Schalters“ die Leber vor langfristigen Schäden schützen?
Ein genauerer Blick darauf, wie die Leber geschädigt wird
Fibrose beginnt, wenn die wichtigsten Funktionszellen der Leber, die Hepatozyten, wiederholt verletzt werden. In dieser Arbeit verwendeten die Forscher ein bewährtes Chemikal, Tetrachlorkohlenstoff, um chronische Leberschäden in Mäusen und in kultivierten Maus-Leberzellen zu imitieren. Die behandelten Mäuse entwickelten Kennzeichen von Lebererkrankungen, wie sie beim Menschen beobachtet werden: erhöhte Blutwerte für Leberschädigung, Fett- und Cholesterinstoffwechselstörungen, Entzündung und ausgeprägte Ablagerungen narbenähnlichen Materials, die die normale Leberarchitektur verzerren. Diese Veränderungen bestätigen, dass das Modell die Abfolge von Ereignissen, die aus einfacher Schädigung dauerhafte Vernarbung entstehen lässt, zuverlässig nachbildet.
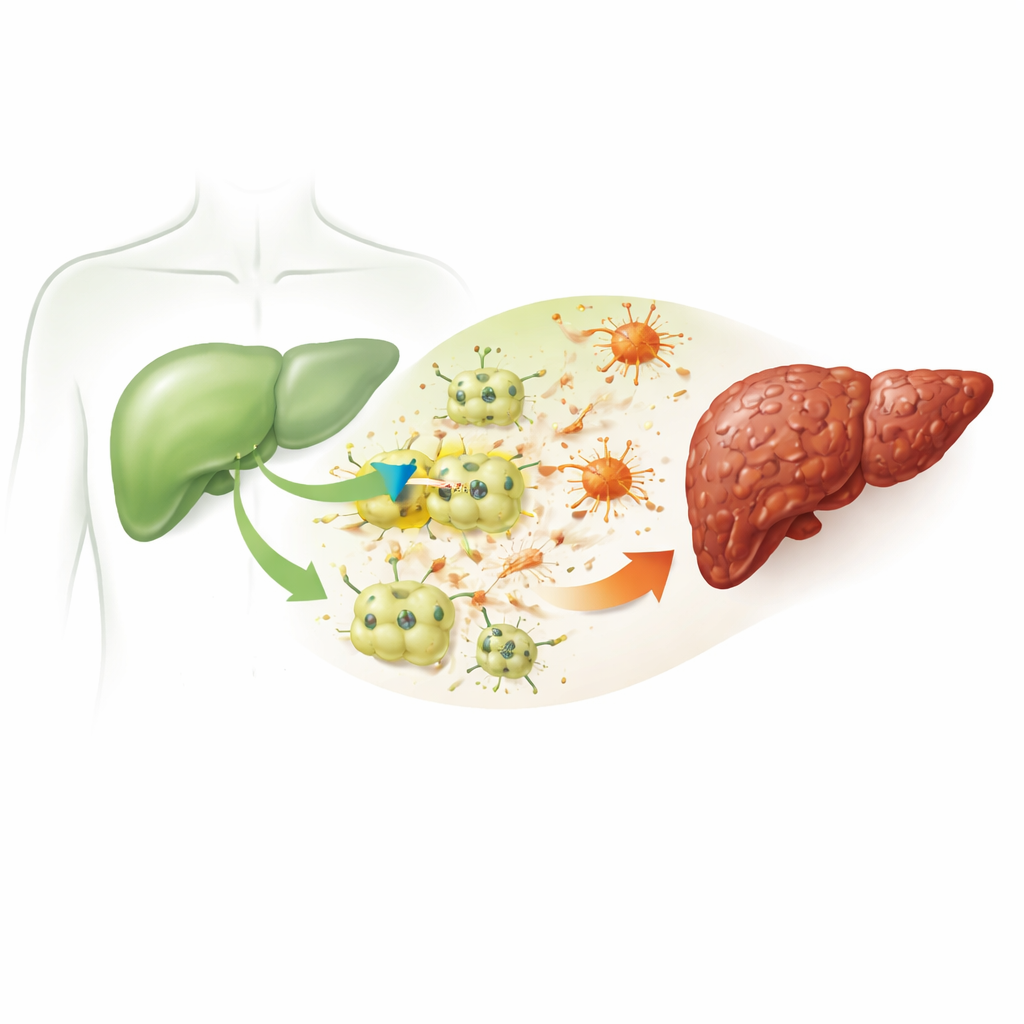
Ein versteckter Kanal, der in kranken Leberzellen auftaucht
Das Team konzentrierte sich auf ein Protein namens ITPR3, einen Kanal, der Calcium aus internen Speichern innerhalb der Zelle freisetzt. Gesunde Hepatozyten stellen diesen Kanal normalerweise überhaupt nicht her. In den geschädigten Mäuselebern und in den chemisch belasteten Leberzellen im Labor fanden die Forscher jedoch einen markanten Anstieg der ITPR3-Spiegel. Dort, wo ITPR3 auftrat, waren Leberzellen deutlich häufiger dem programmierten Zelltod ausgesetzt, einer kontrollierten Form der Selbstvernichtung. Als die Wissenschaftler ein genetisches Werkzeug (siRNA) einsetzten, um ITPR3 herunterzuregulieren, verschwand der Anstieg des intrazellulären Calciums und die Sterberate der Zellen sank deutlich. Die Flüssigkeit aus diesen geschädigten Hepatozyten konnte außerdem benachbarte Stützzellen, sogenannte hepatische Sternzellen, in einen aktiven, narbenproduzierenden Zustand versetzen — eine Veränderung, die abgeschwächt war, wenn ITPR3 unterdrückt wurde.
Vom Calciumsignal zum Entzündungsschalter
Calcium ist nicht nur ein Strukturmineral; innerhalb der Zelle wirken kurze Calcium-Pulse als Signale, die Signalwege ein- und ausschalten. Die Autoren zeigten, dass überschüssiges Calcium, das durch neu gebildete ITPR3-Kanäle freigesetzt wird, einen bekannten Entzündungsregulator, NF-κB, aktiviert. In geschädigten Mäuselebern und in kultivierten Hepatozyten verlegte NF-κB sich vom Zellplasma in den Zellkern, wo Gene gesteuert werden — ein Kennzeichen seiner Aktivierung. Eine Reduktion von ITPR3 verhinderte diese nukleare Verlagerung, während ein chemischer NF-κB-Blocker den Zelltod verringerte. Diese Experimente verbinden das Auftreten von ITPR3 mit einem Calciumanstieg, der Aktivierung von NF-κB und letztlich mit dem Verlust von Hepatozyten, was die Grundlage für Narbenbildung legt.
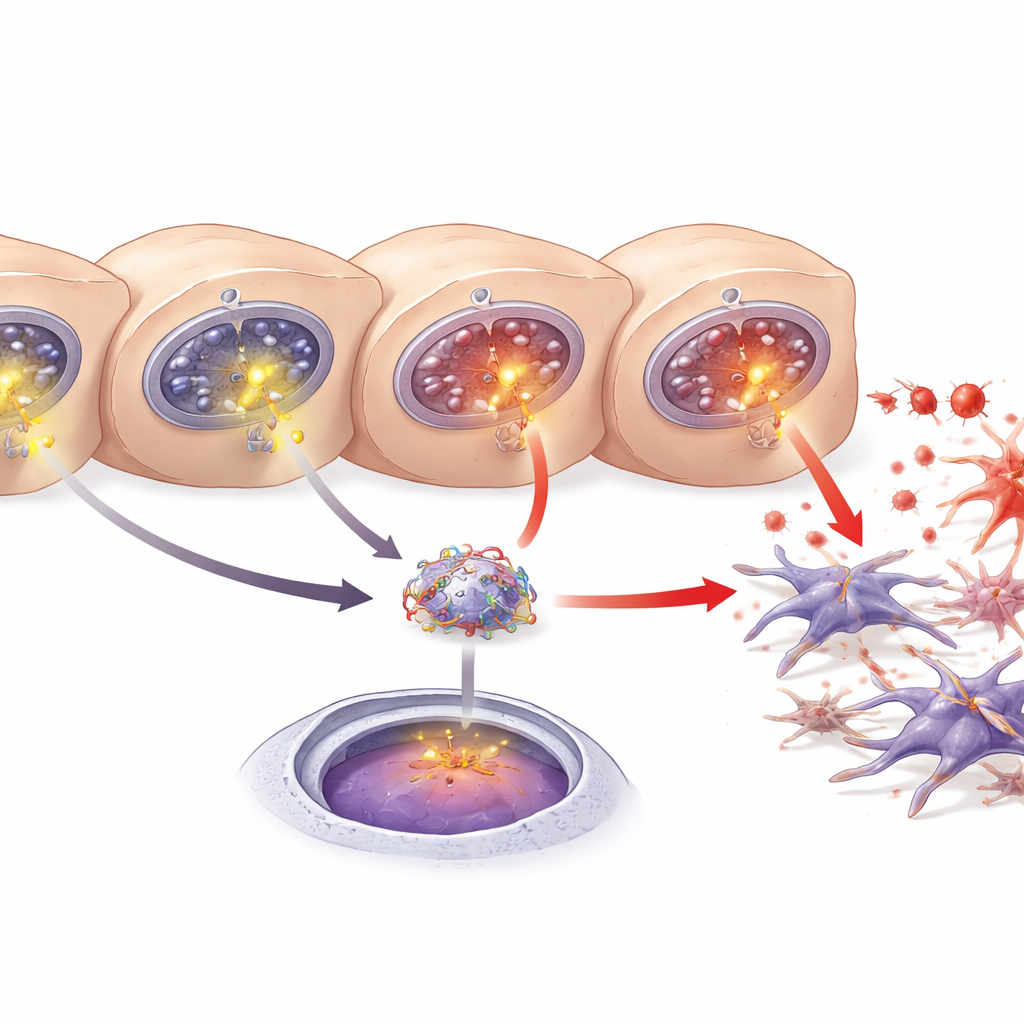
Ein hepatischer Botenstoff, der die Vernarbung antreibt
Ein weiterer Schlüsselakteur ist ein leberabgeleitetes Protein namens LECT2, das zuvor mit Fettlebererkrankung, Fibrose und Leberkrebs in Verbindung gebracht wurde. Die Forscher fanden vermehrt LECT2 im Blut, im Lebergewebe und im Kulturmedium, wenn Hepatozyten geschädigt waren. Mit Hilfe von Datenbankvorhersagen und genetischen Reporter-Tests zeigten sie, dass NF-κB an eine bestimmte DNA-Strecke in der Kontrollregion des LECT2-Gens binden kann und so dessen Aktivität steigert. Die Blockade von NF-κB oder das Herunterregulieren von ITPR3 verringerten sowohl die LECT2-Produktion als auch deren Freisetzung. Zusammengenommen skizzieren die Befunde eine Kette: ITPR3 erscheint in gestressten Hepatozyten, löst vermehrte Calciumfreisetzung aus, aktiviert NF-κB, fördert die LECT2-Produktion und begünstigt so die Aktivierung von Sternzellen und die Fibrose.
Was das für künftige Therapien bedeutet
Einfach gesagt identifiziert die Studie einen abnormalen Calciumkanal, ITPR3, als einen upstream „An-Schalter“, der anhaltende Leberschädigung in dauerhafte Vernarbung verwandelt. Indem gezeigt wird, dass ITPR3 über eine spezifische Calcium–NF-κB–LECT2-Achse zu Zelltod und Fibrose führt, hebt die Arbeit mehrere potenzielle Wirkstoffziele hervor: den Kanal selbst, den Entzündungsschalter oder den nachgeschalteten hepatischen Botenstoff. Zwar sind weitere Untersuchungen am Menschen nötig, insbesondere zur Bestätigung von Sicherheit und klinischem Nutzen, doch deuten die Ergebnisse darauf hin, dass das Abschalten von ITPR3 helfen könnte, Hepatozyten zu erhalten, Entzündungen zu dämpfen und das Fortschreiten chronischer Lebererkrankungen in Richtung Zirrhose zu verlangsamen oder zu verhindern.
Zitation: Zhao, X., Liu, X., Ni, M. et al. ITPR3 promotes liver fibrosis by damaging hepatocytes via the Ca2+/NF-B/LECT2 pathway. Sci Rep 16, 13376 (2026). https://doi.org/10.1038/s41598-026-43866-1
Schlüsselwörter: Leberfibrose, Apoptose von Hepatozyten, Calcium-Signalübertragung, NF-kappaB-Signalweg, LECT2