Clear Sky Science · es
ITPR3 promueve la fibrosis hepática al dañar los hepatocitos a través de la vía Ca2+/NF-κB/LECT2
Por qué importa la cicatrización del hígado
Las enfermedades hepáticas están en aumento en todo el mundo y a menudo progresan silenciosamente durante años antes de que aparezcan los síntomas. Un punto de inflexión clave es la fibrosis hepática, la acumulación de tejido cicatricial que puede llevar finalmente a cirrosis e insuficiencia hepática. Este estudio plantea una pregunta crítica: ¿qué provoca exactamente el daño temprano a las células del hígado que pone en marcha este proceso de cicatrización, y podría bloquearse un único "interruptor" molecular para proteger el hígado del daño a largo plazo?
Una mirada más cercana a cómo se lesiona el hígado
La fibrosis comienza cuando las principales células funcionales del hígado, llamadas hepatocitos, sufren lesiones repetidas. En este estudio, los investigadores usaron un químico bien establecido, el tetracloruro de carbono, para imitar el daño hepático crónico en ratones y en hepatocitos de ratón cultivados. Los ratones tratados desarrollaron signos característicos de enfermedad hepática observados en humanos: elevación de marcadores sanguíneos de lesión hepática, alteraciones en grasas y colesterol, inflamación y abundantes depósitos de material similar a una cicatriz que distorsionan la arquitectura normal del hígado. Estos cambios confirman que el modelo reproduce fielmente la cadena de eventos que convierte una lesión simple en cicatrización duradera.
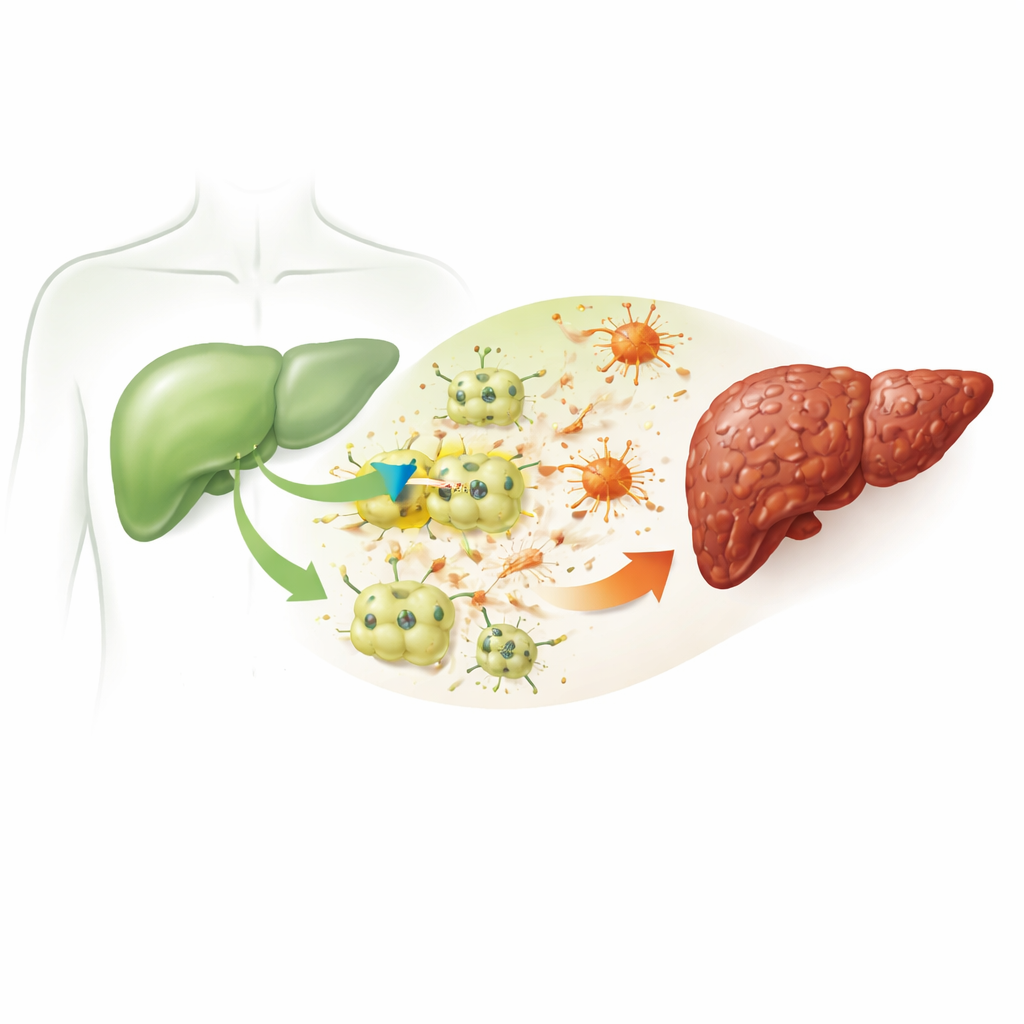
Un canal oculto que aparece en hepatocitos enfermos
El equipo se centró en una proteína llamada ITPR3, un canal que libera calcio desde reservas internas dentro de las células. Los hepatocitos sanos normalmente no producen este canal en absoluto. Sin embargo, tanto en los hígados dañados de los ratones como en las células hepáticas estresadas químicamente cultivadas en el laboratorio, los investigadores encontraron un marcado aumento de los niveles de ITPR3. Donde aparecía ITPR3, las células hepáticas tenían mucha más probabilidad de estar en proceso de muerte programada, una forma controlada de autodestrucción. Cuando los científicos usaron una herramienta genética (siRNA) para reducir ITPR3, el aumento de calcio intracelular desapareció y la tasa de muerte celular disminuyó drásticamente. El fluido procedente de estos hepatocitos dañados también podía transformar a las células de soporte cercanas, llamadas células estrelladas hepáticas, en un estado activo productor de cicatrices—un cambio que se atenuó cuando se suprimió ITPR3.
Del signo de calcio al interruptor inflamatorio
El calcio no es solo un mineral estructural; dentro de las células, pulsos breves de calcio actúan como señales que activan o desactivan vías. Los autores demostraron que el exceso de calcio liberado a través de los canales de ITPR3 recién formados activa un regulador inflamatorio bien conocido, NF‑κB. En los hígados dañados de ratones y en hepatocitos cultivados, NF‑κB se desplazó desde el citoplasma hacia el núcleo, donde se controla la expresión génica, un sello de su activación. Reducir los niveles de ITPR3 impidió este desplazamiento nuclear, mientras que un bloqueador químico de NF‑κB redujo la muerte celular. Estos experimentos vinculan la aparición de ITPR3 con una oleada de calcio, la activación de NF‑κB y, en última instancia, con la pérdida de hepatocitos, lo que prepara el terreno para la formación de cicatrices.
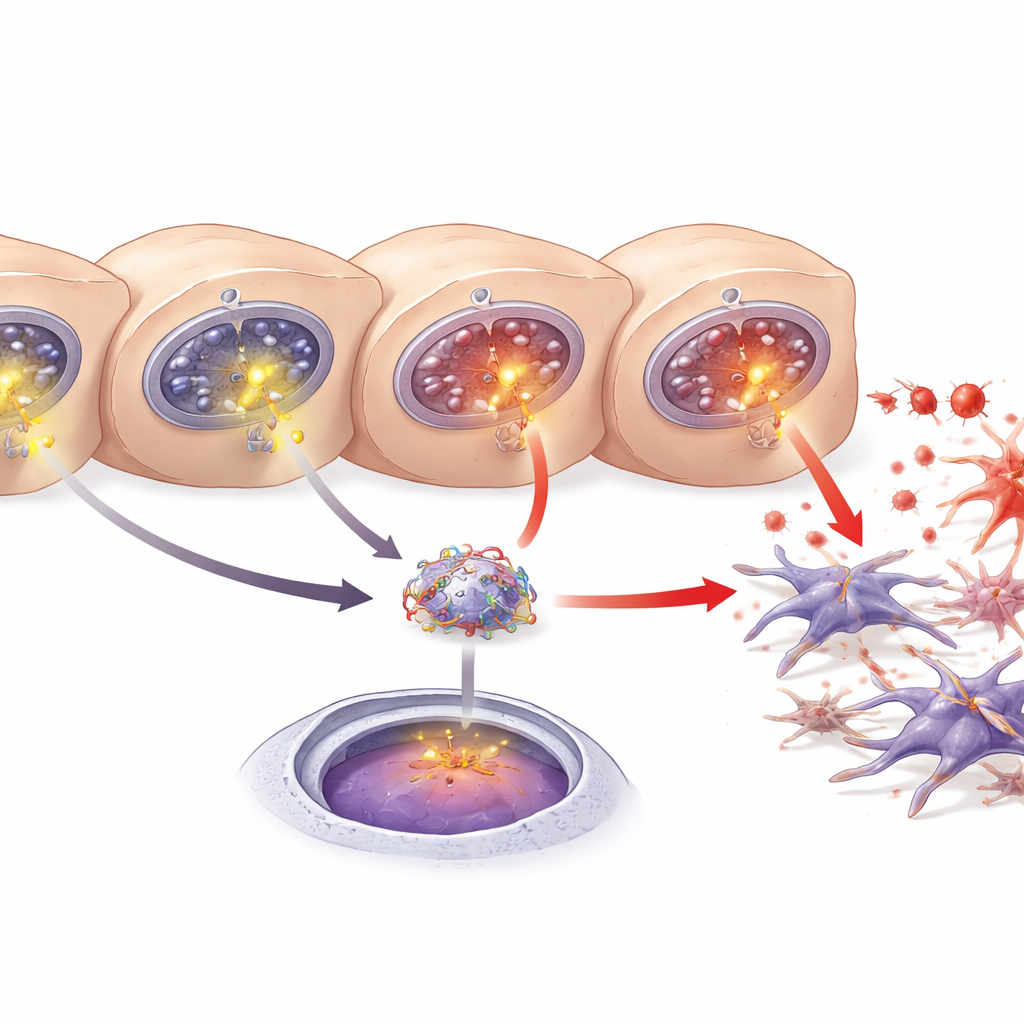
Un mensajero hepático que alimenta la cicatrización
Otro actor clave es una proteína de origen hepático llamada LECT2, previamente implicada en la enfermedad del hígado graso, la fibrosis y el cáncer de hígado. Los investigadores encontraron más LECT2 en la sangre, el tejido hepático y el medio de cultivo cuando los hepatocitos estaban dañados. Usando predicciones de bases de datos y pruebas con reporteros genéticos, mostraron que NF‑κB puede unirse a un tramo específico de ADN en la región reguladora del gen LECT2, aumentando su actividad. Bloquear NF‑κB o reducir ITPR3 disminuyó tanto la producción como la liberación de LECT2. En conjunto, los hallazgos describen una cadena: ITPR3 aparece en hepatocitos estresados, provoca una liberación adicional de calcio, activa NF‑κB, estimula la producción de LECT2 y favorece la activación de las células estrelladas y la fibrosis.
Qué significa esto para futuros tratamientos
En términos sencillos, el estudio identifica un canal de calcio anómalo, ITPR3, como un "interruptor" ascendente que convierte el daño hepático sostenido en cicatrización duradera. Al mostrar que ITPR3 conduce a la muerte celular y a la fibrosis a través de una ruta específica calcio–NF‑κB–LECT2, el trabajo pone de relieve varios posibles objetivos farmacológicos: el propio canal, el interruptor inflamatorio o el mensajero hepático aguas abajo. Aunque se necesita más investigación en humanos, especialmente para confirmar seguridad y beneficios en el mundo real, los resultados sugieren que apagar ITPR3 podría ayudar a preservar los hepatocitos, calmar la inflamación y frenar o prevenir la progresión de la enfermedad hepática crónica hacia la cirrosis.
Cita: Zhao, X., Liu, X., Ni, M. et al. ITPR3 promotes liver fibrosis by damaging hepatocytes via the Ca2+/NF-B/LECT2 pathway. Sci Rep 16, 13376 (2026). https://doi.org/10.1038/s41598-026-43866-1
Palabras clave: fibrosis hepática, apoptosis de hepatocitos, señalización del calcio, vía NF-kappaB, LECT2