Clear Sky Science · en
ITPR3 promotes liver fibrosis by damaging hepatocytes via the Ca2+/NF-B/LECT2 pathway
Why liver scarring matters
Liver disease is rising worldwide, often progressing silently for years before symptoms appear. A key turning point is liver fibrosis, the build‑up of scar tissue that can eventually lead to cirrhosis and liver failure. This study asks a critical question: what exactly drives the earliest damage to liver cells that sets this scarring process in motion, and could blocking a single molecular "switch" help protect the liver from long‑term harm?
A closer look at how the liver gets hurt
Fibrosis begins when the liver’s main working cells, called hepatocytes, are repeatedly injured. In this study, researchers used a well‑established chemical, carbon tetrachloride, to mimic chronic liver damage in mice and in cultured mouse liver cells. The treated mice developed hallmarks of liver disease seen in humans: elevated blood markers of liver injury, fat and cholesterol disturbances, inflammation, and heavy deposits of scar‑like material that distort normal liver architecture. These changes confirm that the model faithfully reproduces the chain of events that turns simple injury into lasting scarring.
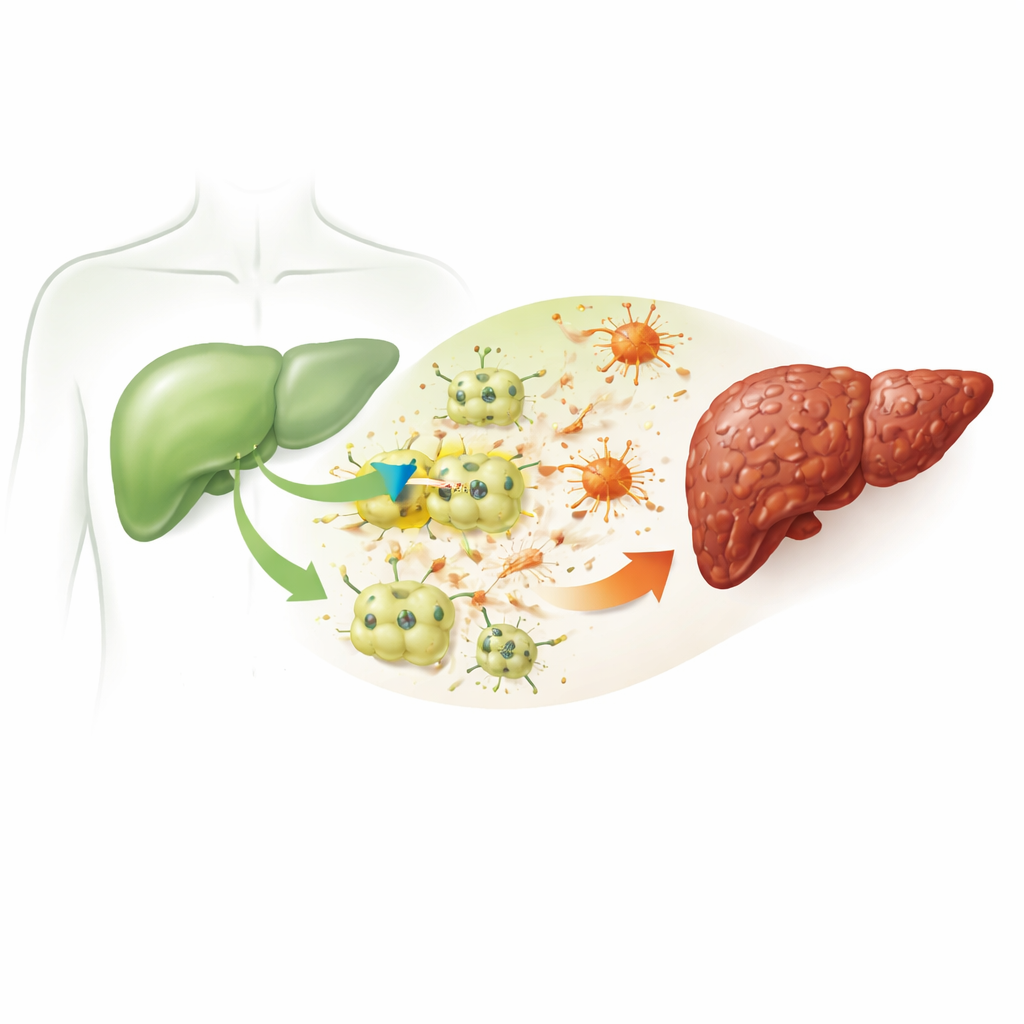
A hidden channel that appears in sick liver cells
The team focused on a protein called ITPR3, a channel that releases calcium from internal stores inside cells. Healthy hepatocytes normally do not make this channel at all. However, in both the damaged mouse livers and the chemically stressed liver cells grown in the lab, the researchers found a striking rise in ITPR3 levels. Where ITPR3 appeared, liver cells were much more likely to be undergoing programmed cell death, a controlled form of self‑destruction. When the scientists used a genetic tool (siRNA) to dial down ITPR3, the surge in internal calcium disappeared, and the rate of cell death dropped sharply. Fluid from these damaged hepatocytes could also switch nearby support cells, called hepatic stellate cells, into an active, scar‑producing state—a change that was blunted when ITPR3 was suppressed.
From calcium signal to inflammatory switch
Calcium is not just a structural mineral; inside cells, brief calcium pulses act as signals that turn pathways on and off. The authors showed that excess calcium released through newly formed ITPR3 channels flips on a well‑known inflammatory regulator, NF‑κB. In damaged mouse livers and cultured hepatocytes, NF‑κB shifted from the cell fluid into the nucleus, where genes are controlled, a hallmark of its activation. Reducing ITPR3 levels prevented this nuclear shift, while a chemical NF‑κB blocker reduced cell death. These experiments link the appearance of ITPR3 to a calcium surge, activation of NF‑κB, and ultimately to loss of hepatocytes, setting the stage for scar formation.
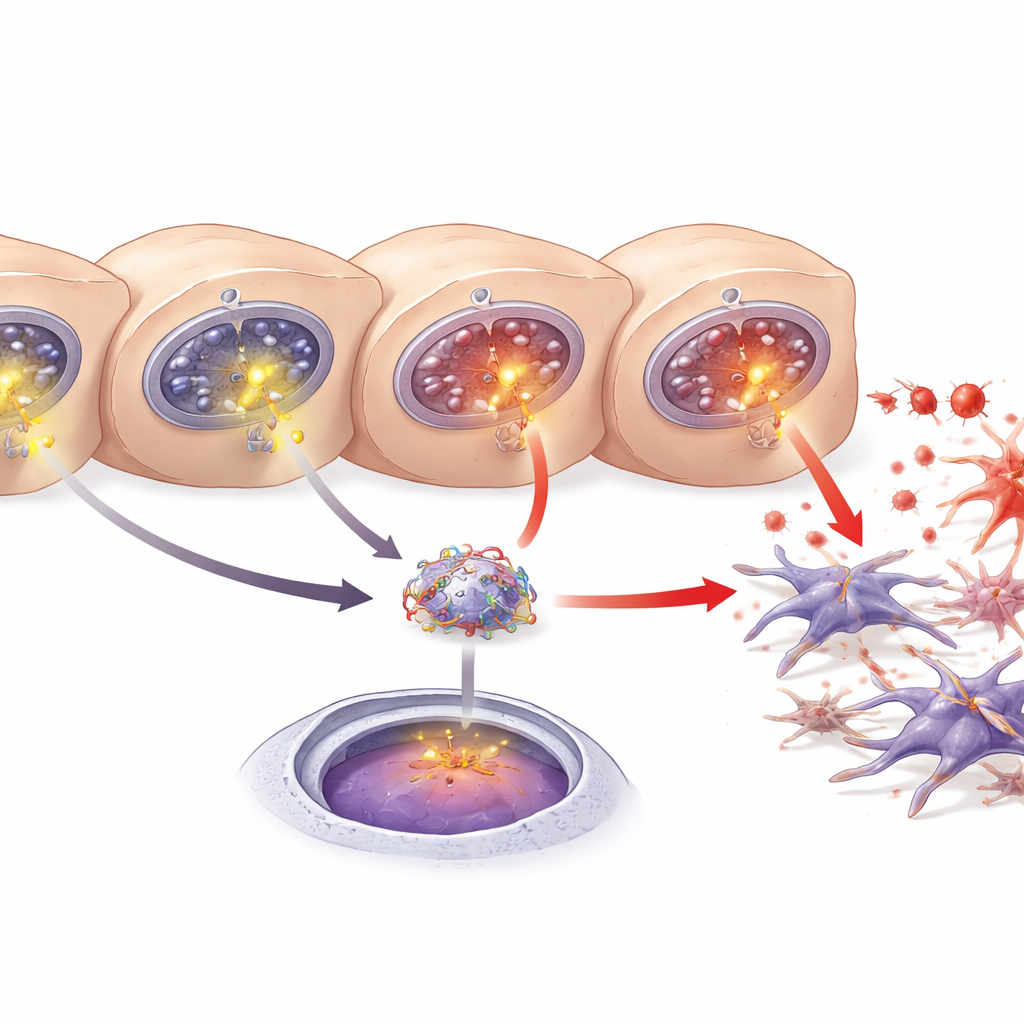
A liver messenger that fuels scarring
Another key player is a liver‑derived protein called LECT2, previously implicated in fatty liver disease, fibrosis, and liver cancer. The researchers found more LECT2 in the blood, liver tissue, and culture media when hepatocytes were damaged. Using database predictions and genetic reporter tests, they showed that NF‑κB can latch onto a specific stretch of DNA in the LECT2 gene’s control region, boosting its activity. Blocking NF‑κB or knocking down ITPR3 both reduced LECT2 production and release. Together, the findings outline a chain: ITPR3 appears in stressed hepatocytes, triggers extra calcium release, activates NF‑κB, drives LECT2 production, and promotes stellate‑cell activation and fibrosis.
What this means for future treatments
In plain terms, the study identifies an abnormal calcium channel, ITPR3, as an upstream "on switch" that converts ongoing liver injury into lasting scarring. By showing that ITPR3 leads to cell death and fibrosis through a specific calcium–NF‑κB–LECT2 route, the work highlights several potential drug targets: the channel itself, the inflammatory switch, or the downstream liver messenger. While more research is needed in people, especially to confirm safety and real‑world benefits, the results suggest that turning off ITPR3 could help preserve liver cells, calm inflammation, and slow or prevent the progression of chronic liver disease toward cirrhosis.
Citation: Zhao, X., Liu, X., Ni, M. et al. ITPR3 promotes liver fibrosis by damaging hepatocytes via the Ca2+/NF-B/LECT2 pathway. Sci Rep 16, 13376 (2026). https://doi.org/10.1038/s41598-026-43866-1
Keywords: liver fibrosis, hepatocyte apoptosis, calcium signaling, NF-kappaB pathway, LECT2