Clear Sky Science · ar
ITPR3 يعزز تليف الكبد عن طريق إتلاف الخلايا الكبدية عبر مسار Ca2+/NF-B/LECT2
لماذا يهم تندب الكبد
أمراض الكبد في ازدياد على مستوى العالم، وغالباً ما تتفاقم بصمت لسنوات قبل ظهور الأعراض. نقطة التحول المهمة هي تليف الكبد، أي تراكم النسيج الندبي الذي قد يؤدي في النهاية إلى تشمع الكبد وفشل كبدي. تطرح هذه الدراسة سؤالاً حاسماً: ما الذي يسبب الضرر المبكر للخلايا الكبدية الذي يشغل عملية التندب هذه، وهل يمكن أن يساعد حظر «مفتاح» جزيئي واحد في حماية الكبد من الأذى طويل الأمد؟
نظرة أقرب على كيفية إصابة الكبد
يبدأ التليف عندما تتعرض الخلايا الكبدية الرئيسة، المسماة بالهرباتوسايتات، لإصابات متكررة. في هذه الدراسة، استخدم الباحثون مادة كيميائية معروفة جيداً، رباعي كلورو الكربون، لمحاكاة الضرر الكبدي المزمن في الفئران وفي خلايا كبدية مأخوذة من الفئران ومعزولة في المختبر. طورت الفئران المعالجة علامات مميزة لمرض الكبد تُرى لدى البشر: ارتفاع مؤشرات إصابة الكبد في الدم، اضطرابات في الدهون والكوليسترول، التهاب، وترسبات كثيفة من مادة شبيهة بالندبة تشوّه البنية الطبيعية للكبد. تؤكد هذه التغيرات أن النموذج يعيد بدقة سلسلة الأحداث التي تحول إصابة بسيطة إلى تندب مستمر.
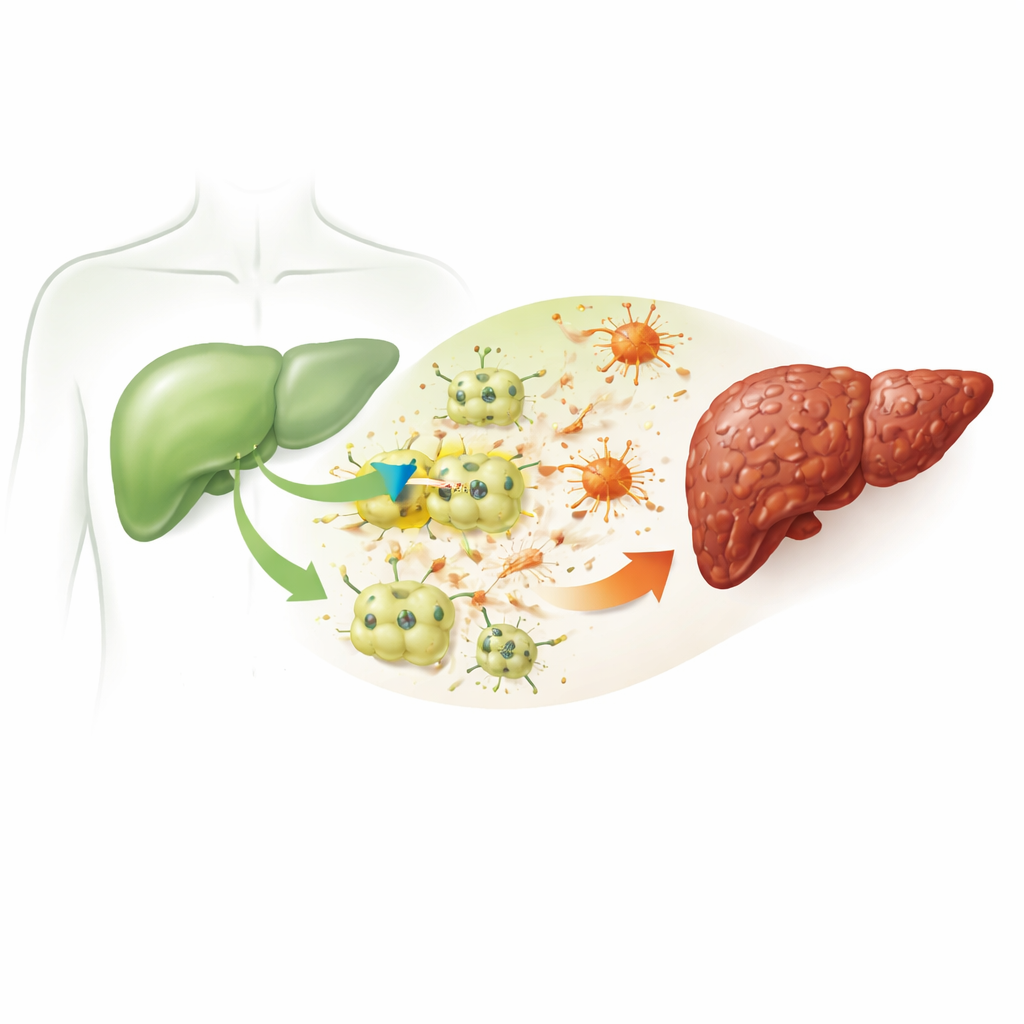
قناة مخفية تظهر في الخلايا الكبدية المريضة
ركز الفريق على بروتين يسمى ITPR3، وهو قناة تطلق الكالسيوم من المخازن الداخلية داخل الخلايا. الخلايا الكبدية السليمة عادة لا تصنع هذه القناة على الإطلاق. ومع ذلك، في كل من كبد الفئران المتضرر والخلايا الكبدية المعرضة للضغط الكيميائي في المختبر، وجد الباحثون ارتفاعاً لافتاً في مستويات ITPR3. حيثما ظهرت ITPR3، كانت الخلايا الكبدية أكثر عرضة لأن تخضع للموت المبرمج للخلايا، وهو شكل من أشكال الانتحار الخلوي المنضبط. عندما استخدم العلماء أداة جينية (siRNA) لتقليل ITPR3، اختفى الارتفاع في الكالسيوم الداخلي، وانخفضت نسبة موت الخلايا بشكل كبير. كذلك ساعد السائل الخارج من هذه الخلايا الكبدية المتضررة على تحويل الخلايا الداعمة المجاورة، المسماة بالخلايا الخطافية الكبدية، إلى حالة نشطة منتجة للندب — وهو تغير تقلص عندما تم قمع ITPR3.
من إشارة الكالسيوم إلى مفتاح الالتهاب
الكالسيوم ليس معدناً بنيوياً فحسب؛ داخل الخلايا تعمل نبضات الكالسيوم القصيرة كإشارات تشغل أو تطفئ المسارات الخلوية. أظهر المؤلفون أن الكالسيوم الزائد المنطلق عبر قنوات ITPR3 المتشكلة حديثاً يشغل منظم الالتهاب المعروف NF‑κB. في كبد الفئران المتضرر والهرباتوسايتات المستزرعة، تحرك NF‑κB من سائل الخلية إلى نواة الخلية، حيث تُدار الجينات، وهو سمة من سمات تنشيطه. وبخفض مستويات ITPR3 منعوا هذا الانتقال إلى النواة، بينما قلّل مُثبط كيميائي لـ NF‑κB من موت الخلايا. تربط هذه التجارب بين ظهور ITPR3، وارتفاع الكالسيوم، وتنشيط NF‑κB، وفي النهاية بفقدان الخلايا الكبدية الذي يمهد الطريق لتكوين الندبات.
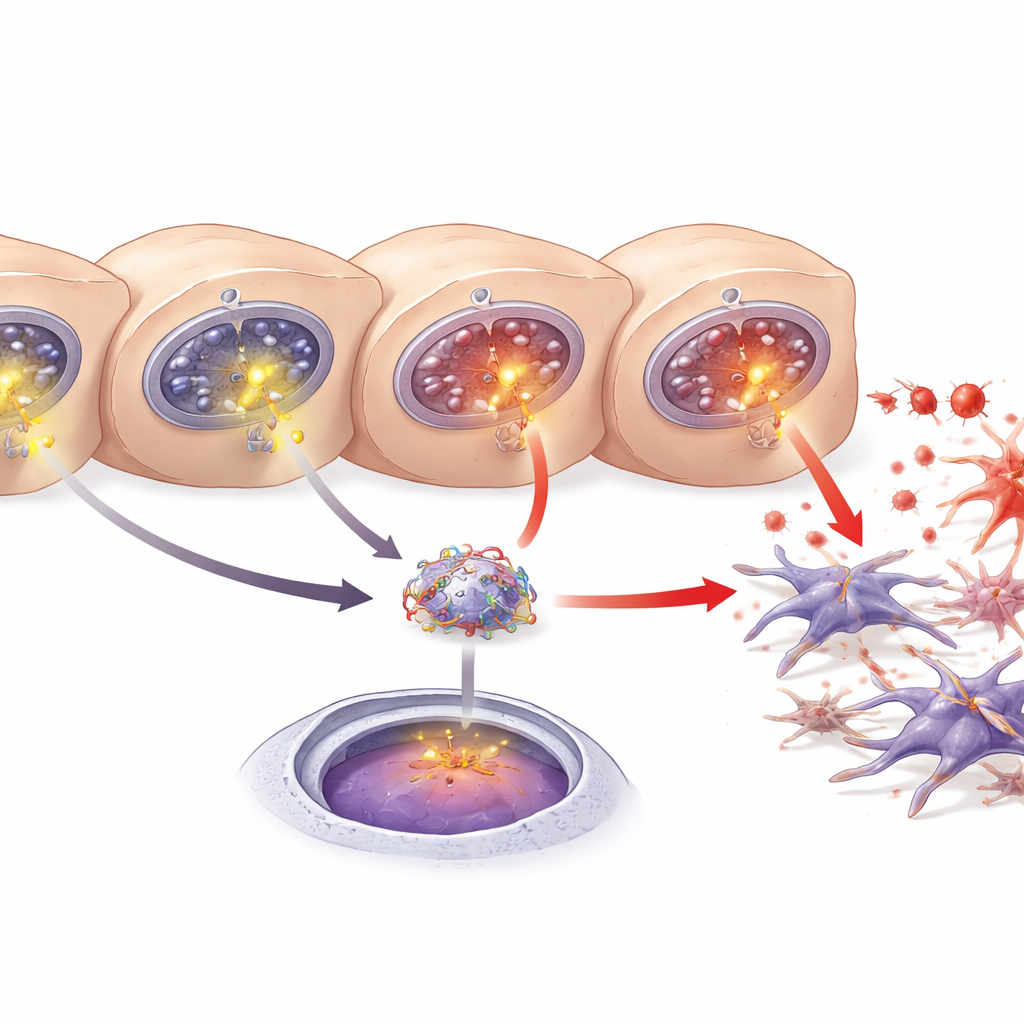
رسل كبدي يغذي التندب
لاعب رئيسي آخر هو بروتين منشأه الكبد يدعى LECT2، الذي ارتبط سابقاً بمرض الكبد الدهني والتليف وسرطان الكبد. وجد الباحثون مستويات أعلى من LECT2 في الدم ونسيج الكبد ووسط الاستزراع عندما تعرضت الخلايا الكبدية للضرر. باستخدام توقعات قواعد بيانات واختبارات ريبورتير جينية، أظهروا أن NF‑κB يمكنه الالتحام بمقطع محدد من الحمض النووي في منطقة التحكم بجين LECT2، معزّزاً نشاطه. أدى حظر NF‑κB أو خفض ITPR3 إلى تقليل إنتاج LECT2 وإفرازه. مجتمعة، توضح النتائج سلسلة: يظهر ITPR3 في الهرباتوسايتات المتوترة، يسبب إفرازاً إضافياً للكالسيوم، ينشط NF‑κB، يحفز إنتاج LECT2، ويعزز تنشيط الخلايا الخطافية والتليف.
ما معنى هذا للعلاجات المستقبلية
بعبارات بسيطة، تحدد الدراسة قناة كلسية غير طبيعية، ITPR3، باعتبارها «مفتاح تشغيل» علوي يحول الإصابة الكبدية المستمرة إلى تندب دائم. من خلال إظهار أن ITPR3 يقود إلى موت الخلايا والتليف عبر مسار كلسي–NF‑κB–LECT2 محدد، تُبرز الدراسة عدة أهداف دوائية محتملة: القناة نفسها، مفتاح الالتهاب، أو الرسول الكبدي المتبع. على الرغم من الحاجة لمزيد من الأبحاث على البشر، لا سيما لتأكيد السلامة والفوائد في العالم الحقيقي، تشير النتائج إلى أن إيقاف ITPR3 قد يساعد في حفظ الخلايا الكبدية، وتهدئة الالتهاب، وإبطاء أو منع تقدم مرض الكبد المزمن نحو التشمع.
الاستشهاد: Zhao, X., Liu, X., Ni, M. et al. ITPR3 promotes liver fibrosis by damaging hepatocytes via the Ca2+/NF-B/LECT2 pathway. Sci Rep 16, 13376 (2026). https://doi.org/10.1038/s41598-026-43866-1
الكلمات المفتاحية: تليف الكبد, استماتة الخلايا الكبدية, إشارة الكالسيوم, مسار NF-kappaB, LECT2