Clear Sky Science · zh
将替代性碎裂技术整合到标准液相色谱-质谱工作流程中:使用单一深度学习模型提升蛋白质组覆盖度
看见更多生命中的蛋白机器
你体内的每个细胞都充满了数千种不同的蛋白质,各自执行特定任务。现代质谱通过将蛋白质切分并称量碎片,已经能读出许多这些蛋白,但重要的部分仍然不可见——尤其是不寻常的蛋白形式以及驱动健康与疾病的微妙化学修饰。本研究描述了一种新方法,将若干先进的碎裂手段与单一人工智能模型结合,使科学家在常规实验中能看到更多的蛋白世界。
蛋白通常如何被“读取”
在大多数实验室中,蛋白质首先被切成称为肽的小片段,然后进入分离并称重的仪器。为了确定每个肽的序列,仪器会故意将这些片段撞碎并记录碎片模式,类似于把花瓶打碎后从碎片推断形状。多年来,基于碰撞的方法——通过肽与气体分子碰撞将其破裂——一直是主力,因为它快速、稳健并且有良好的软件支持。然而,这种标准方法难以保留敏感的化学标记,且会错过复杂蛋白形式的某些部分,从而在我们对生物学的理解中留下盲点。
新的蛋白断裂方式
研究人员开发了其他裂解肽的方法:使用紫外光或电子束,这些方法沿不同路径切割蛋白并且常常保留脆弱特征。这些方法能产生更丰富、更具信息量的碎片模式,但速度较慢、技术要求高且数据分析工具支持不足。为了解决这一问题,作者基于一种专门的质谱仪,能够在一个平台上并在标准液相色谱–质谱工作流程所需的时间尺度内应用碰撞、电子和光子三种碎裂方法。他们为每种方法仔细调校操作条件——例如激光能量或电子暴露时间——以便从复杂的人类细胞样本中获得尽可能多的有用谱图。
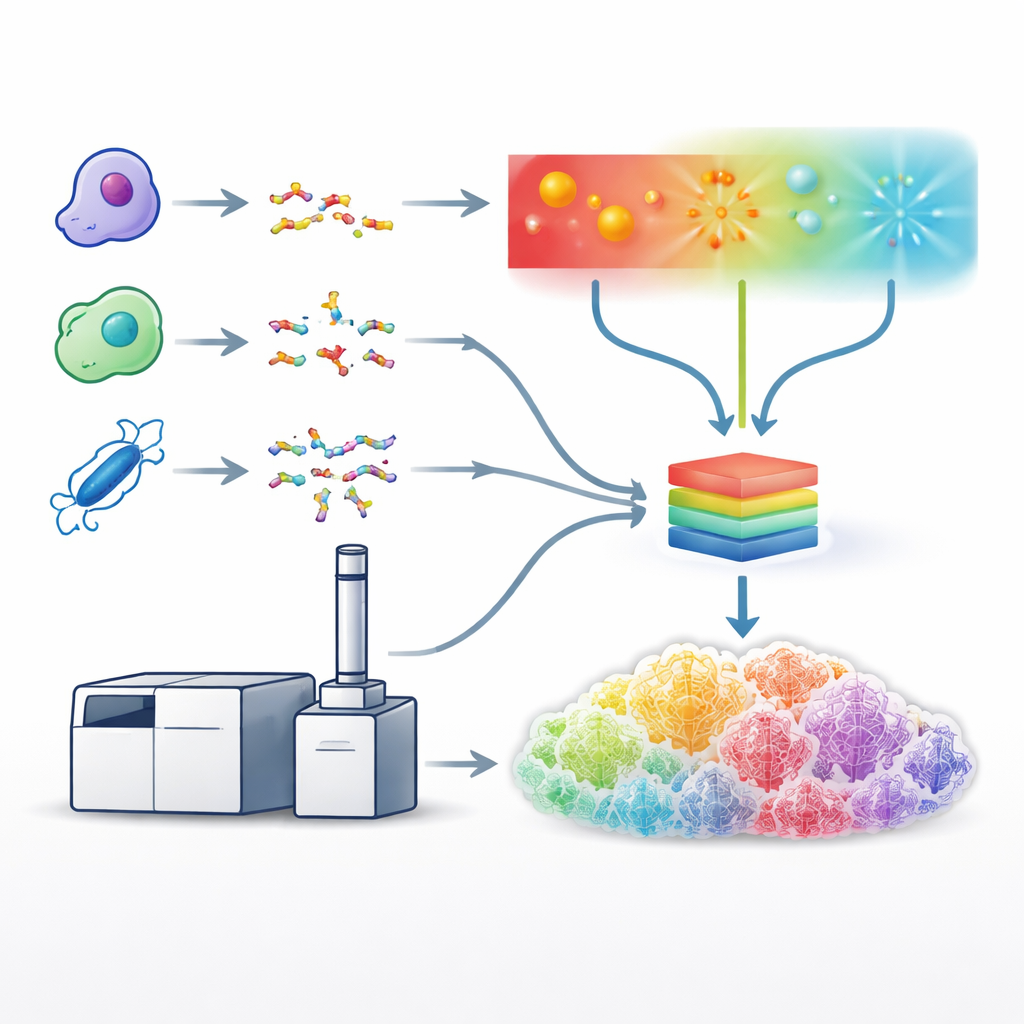
构建统一的学习模型
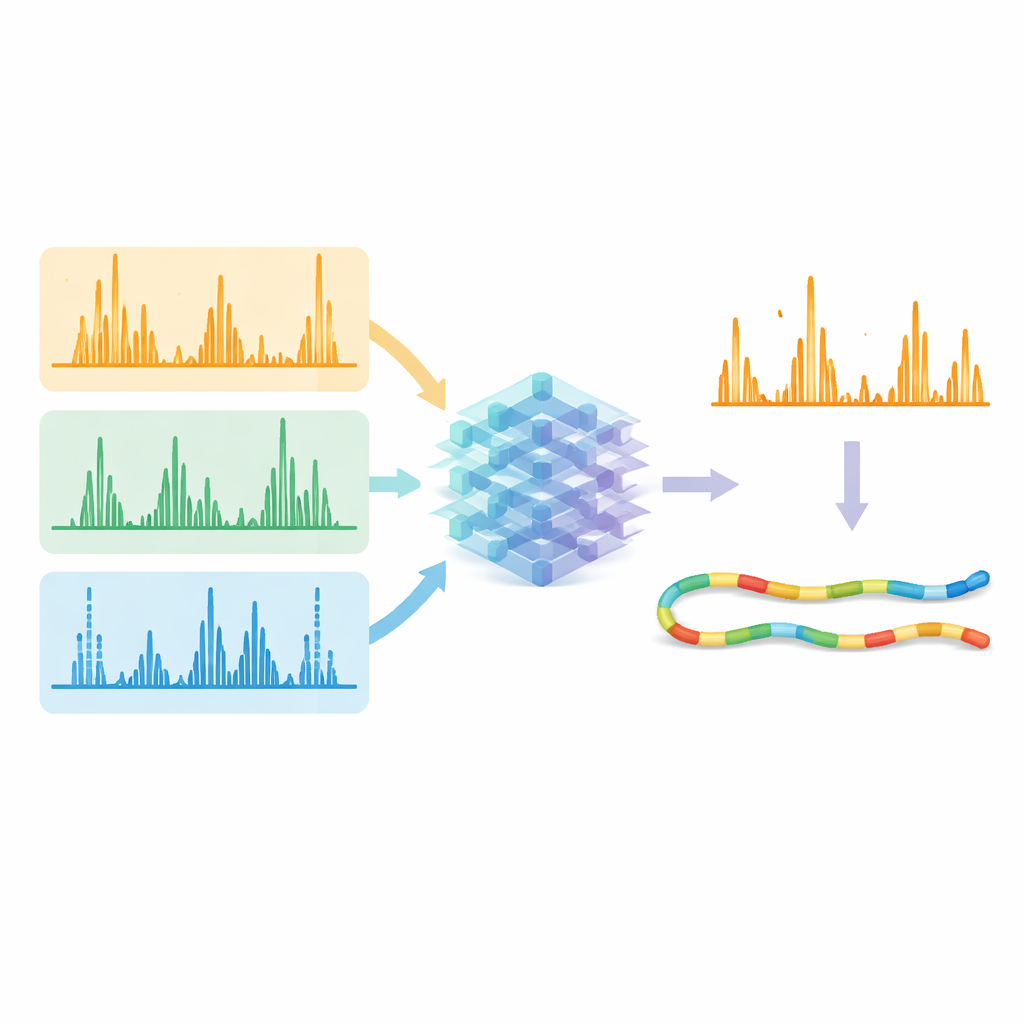
在这些优化方法就绪后,团队使用五种不同的切割酶生成了大量数据集,带来了丰富多样的肽序列。随后,他们利用这些数据训练了一个单一的深度学习模型,这是名为Prosit系统的增强版本,用以同时预测所有碎裂类型的碎片峰的详细模式与强度。模型并非将每种方法单独处理,而是以肽序列、其电荷和所用的断裂方法作为输入,输出数百种可能碎片类型的预期强度。预测谱图与实验数据在各方法间非常吻合,表明模型已有效学习到光、电子和碰撞断裂产生的特征“指纹”。
让人工智能提纯信号
真正的考验在于这些预测能否提升从原始数据中可靠识别肽的数量。研究人员将实测谱图与AI预测谱同时送入现有的搜索与重新评分工具。当软件集中于模型预测应当强且存在的碎片时,正确匹配比错误匹配更易脱颖而出。在不同碎裂方法和酶产生的数据中,可信识别的肽-谱图匹配数通常提升超过10%,在一些具有挑战性的情况下甚至超过30%。重要的是,使用电子和紫外光的替代方法现在在识别效率上达到了与标准碰撞方法相当的水平,同时提供了更广的序列覆盖和关于蛋白的更多独特信息。
将先进方法带入日常应用
由于该AI模型是免费提供并已集成到流行的质谱软件中,它不仅可用于传统的靶向测量,也可用于较新的数据独立采集策略,这些策略一次扫描样品的广阔区域。对人类、植物和细菌细胞混合样本的测试显示,该模型在物种间具有良好的泛化性。就实际意义而言,这项工作消除了将强大但未被充分利用的碎裂方法限制在专家手中的关键障碍。通过在一个预测模型下统一不同的蛋白断裂方式,该研究为常规、高覆盖度的复杂蛋白景观分析提供了路径,使研究人员更容易发现稀有变体、绘制修饰图谱,并最终更完整地理解蛋白在健康与疾病中的行为。
引用: Levin, N., Saylan, C.C., Lapin, J. et al. Integration of alternative fragmentation techniques into standard LC-MS workflows using a single deep learning model enhances proteome coverage. Nat Methods 23, 805–814 (2026). https://doi.org/10.1038/s41592-026-03042-9
关键词: 蛋白质组学, 质谱, 深度学习, 蛋白质断裂, 谱图预测