Clear Sky Science · fr
Intégration de techniques alternatives de fragmentation dans les flux LC‑MS standard via un seul modèle d’apprentissage profond améliore la couverture du protéome
Voir davantage de la machinerie protéique de la vie
Chaque cellule de votre corps contient des milliers de protéines différentes, chacune exécutant une tâche précise. La spectrométrie de masse moderne peut déjà lire beaucoup de ces protéines en les fragmentant puis en pesant les fragments, mais des éléments importants restent invisibles — en particulier des formes protéiques inhabituelles et des modifications chimiques subtiles qui gouvernent la santé et la maladie. Cette étude décrit une nouvelle manière de combiner plusieurs méthodes avancées de fragmentation avec un seul modèle d’intelligence artificielle afin que les scientifiques puissent voir beaucoup plus du monde protéique dans une expérience de routine.
Comment on lit habituellement les protéines
Dans la plupart des laboratoires, les protéines sont d’abord découpées en morceaux plus petits appelés peptides, puis injectées dans un instrument qui les sépare et les pèse. Pour déterminer la séquence de chaque peptide, l’instrument les casse délibérément et enregistre le motif des fragments, comme briser un vase et déduire sa forme à partir des éclats. Pendant des années, une méthode basée sur les collisions — où les peptides sont fragmentés en percutant des molécules de gaz — a été l’approche dominante parce qu’elle est rapide, robuste et bien prise en charge par les logiciels. Cependant, cette méthode standard peine à préserver des étiquettes chimiques délicates et manque des portions de formes protéiques complexes, laissant des angles morts dans notre compréhension de la biologie.
Nouvelles façons de briser les protéines
Les chercheurs ont développé d’autres manières de fissurer les peptides : en utilisant de la lumière ultraviolette ou des faisceaux d’électrons, qui coupent les protéines selon des trajectoires différentes et préservent souvent des caractéristiques fragiles. Ces approches peuvent générer des motifs de fragments plus riches et plus informatifs, mais elles sont plus lentes, techniquement exigeantes et peu prises en charge par les outils d’analyse de données. Pour répondre à ce défi, les auteurs se sont appuyés sur un spectromètre de masse spécialisé capable d’appliquer sur une même plateforme des méthodes de fragmentation par collision, électron et photon, et ce à l’échelle de temps requise pour les flux LC‑MS standard. Ils ont soigneusement ajusté les conditions opératoires de chaque méthode — telles que l’énergie du laser ou le temps d’exposition aux électrons — afin que chacune produise le maximum de spectres utiles à partir d’échantillons cellulaires humains complexes.
Construction d’un modèle d’apprentissage unifié
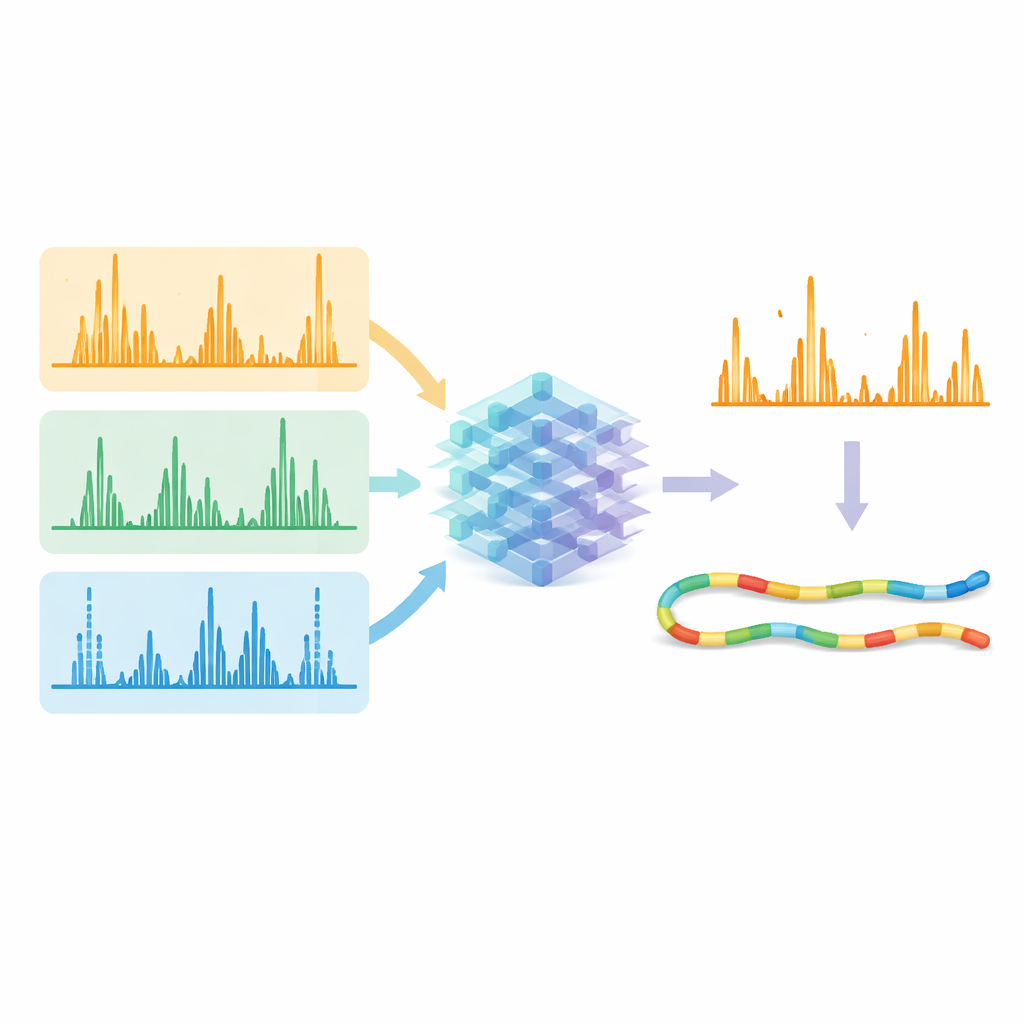
Avec ces méthodes optimisées en place, l’équipe a généré d’immenses jeux de données en utilisant cinq enzymes différentes de découpe des protéines, ce qui a produit une grande diversité de séquences peptidiques. Ils ont ensuite utilisé ces jeux de données pour entraîner un seul modèle d’apprentissage profond, une version améliorée d’un système appelé Prosit, afin de prédire le motif détaillé et l’intensité des pics de fragments pour tous les types de fragmentation simultanément. Plutôt que de traiter chaque méthode séparément, le modèle prend en entrée la séquence peptidique, sa charge et la méthode de fragmentation utilisée, et renvoie les intensités attendues pour des centaines de types de fragments possibles. Les spectres prédits correspondaient très bien aux données expérimentales à travers les méthodes, montrant que le modèle avait effectivement appris les « empreintes » caractéristiques produites par les fragmentations par lumière, électrons et collisions.
Laisser l’IA nettoyer le signal
Le véritable test était de savoir si ces prédictions pouvaient améliorer le nombre de peptides identifiés de façon fiable à partir des données brutes. Les chercheurs ont alimenté à la fois les spectres mesurés et les motifs prédits par l’IA dans des outils existants de recherche et de rescoring. En demandant au logiciel de se concentrer sur les fragments que le modèle prédisait comme devant être intenses et présents, les correspondances correctes ressortaient plus clairement des fausses. Sur des données collectées avec différentes méthodes de fragmentation et enzymes, le nombre d’accords peptide–spectre identifiés de manière confiante augmentait typiquement de plus de 10 %, et dans certains cas difficiles de plus de 30 %. Fait important, les méthodes alternatives utilisant des électrons et la lumière ultraviolette atteignaient désormais une efficacité d’identification similaire à celle de la méthode par collision standard, tout en fournissant une couverture de séquence plus large et des informations plus uniques sur les protéines.
Intégrer les méthodes avancées dans l’usage courant
Parce que le modèle d’IA est librement disponible et intégré aux logiciels de spectrométrie de masse populaires, il peut être utilisé non seulement pour des mesures traditionnelles ciblées mais aussi pour de nouvelles stratégies d’acquisition indépendante des données qui balaient de larges portions de l’échantillon à la fois. Des tests sur des mélanges de cellules humaines, végétales et bactériennes ont montré que le modèle se généralise bien entre les espèces. En termes pratiques, ce travail supprime un obstacle clé qui confinait auparavant des méthodes de fragmentation puissantes mais sous‑utilisées aux spécialistes. En unifiant différentes façons de fragmenter les protéines sous un modèle prédictif unique, l’étude ouvre la voie à des analyses de routine à haute couverture des paysages protéiques complexes, facilitant la détection de variants rares, la cartographie des modifications et, au final, une compréhension plus complète du comportement des protéines en santé et en maladie.
Citation: Levin, N., Saylan, C.C., Lapin, J. et al. Integration of alternative fragmentation techniques into standard LC-MS workflows using a single deep learning model enhances proteome coverage. Nat Methods 23, 805–814 (2026). https://doi.org/10.1038/s41592-026-03042-9
Mots-clés: protéomique, spectrométrie de masse, apprentissage profond, fragmentation des protéines, prédiction spectrale