Clear Sky Science · en
Integration of alternative fragmentation techniques into standard LC-MS workflows using a single deep learning model enhances proteome coverage
Seeing More of Life’s Protein Machinery
Every cell in your body is packed with thousands of different proteins, each carrying out a specific task. Modern mass spectrometry can already read many of these proteins by breaking them into pieces and weighing the fragments, but important parts still remain invisible—especially unusual protein forms and subtle chemical tweaks that drive health and disease. This study describes a new way to combine several advanced fragmentation methods with a single artificial intelligence model so that scientists can see much more of the protein world in a routine experiment.
How Proteins Are Usually Read
In most labs, proteins are first chopped into smaller pieces called peptides and then fed into an instrument that separates and weighs them. To figure out each peptide’s sequence, the instrument deliberately smashes these pieces and records the pattern of fragments, like shattering a vase and inferring its shape from the shards. For years, a collision-based method—where peptides are broken by bumping into gas molecules—has been the workhorse because it is fast, robust and well supported by software. However, this standard approach struggles to keep delicate chemical tags on the protein intact and misses parts of complex protein forms, leaving blind spots in our understanding of biology.
New Ways to Break Proteins Apart
Researchers have developed other ways to crack peptides: using ultraviolet light, or beams of electrons, which slice proteins along different paths and often preserve fragile features. These approaches can generate richer and more informative fragment patterns, but they are slower, technically demanding and poorly supported by data analysis tools. To tackle this, the authors built on a specialized mass spectrometer that can apply collision-, electron- and photon-based breaking methods in one platform and on the time scale needed for standard liquid chromatography–mass spectrometry workflows. They carefully tuned the operating conditions for each method—such as laser energy or electron exposure time—so that each produced as many useful spectra as possible from complex human cell samples.
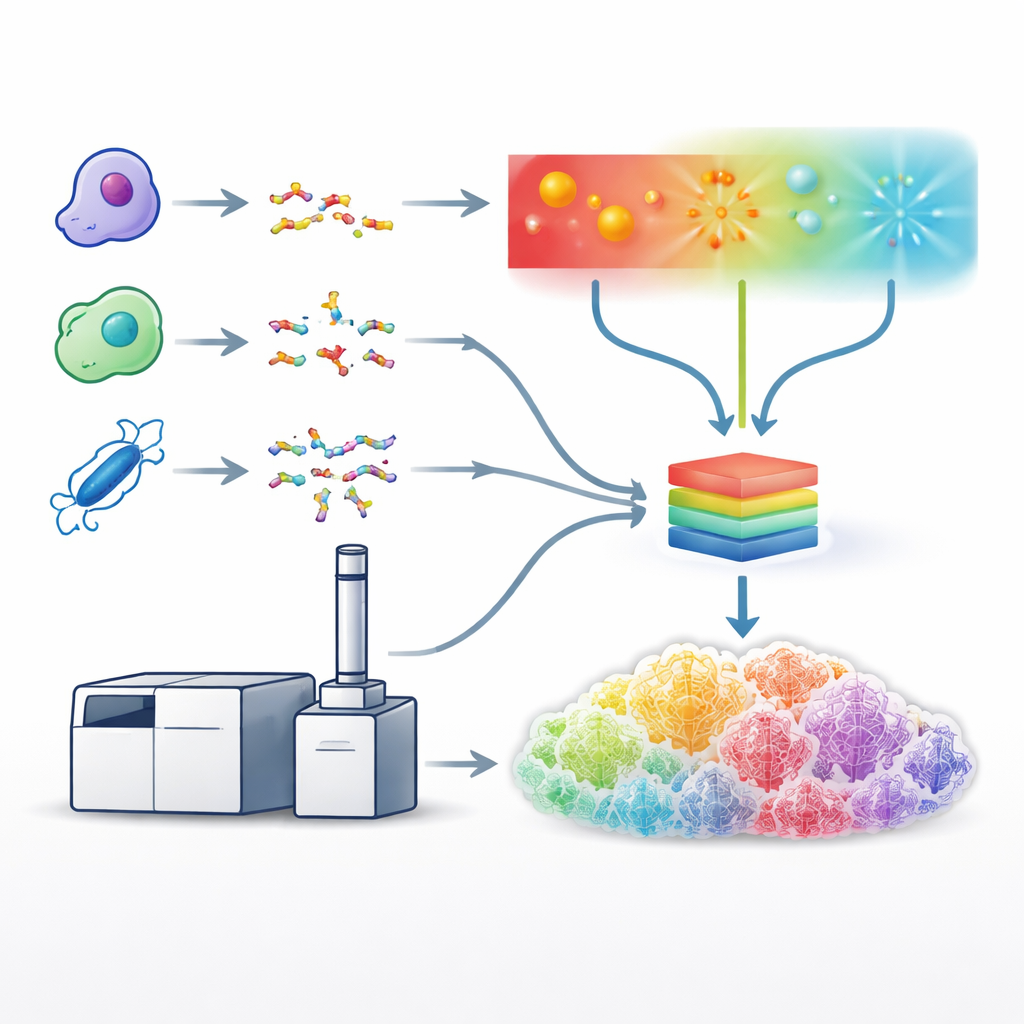
Building a Unified Learning Model
With these optimized methods in place, the team generated vast datasets using five different protein-cutting enzymes, which yielded a huge diversity of peptide sequences. They then used these datasets to train a single deep learning model, an enhanced version of a system called Prosit, to predict the detailed pattern and intensity of fragment peaks for all fragmentation types at once. Instead of treating each method separately, the model takes as input the peptide sequence, its charge and which breaking method was used, and outputs the expected intensities for hundreds of possible fragment types. The predicted spectra matched experimental data very closely across methods, showing that the model had effectively learned the characteristic “fingerprints” produced by light-, electron- and collision-based breaking.
Letting AI Clean Up the Signal
The real test was whether these predictions could improve how many peptides are confidently identified from raw data. The researchers fed both the measured spectra and the AI-predicted patterns into existing search and rescoring tools. When they asked the software to focus on fragments that the model said should be strong and present, correct matches stood out more clearly from false ones. Across data collected by different fragmentation methods and enzymes, the number of confidently identified peptide–spectrum matches typically rose by more than 10%, and in some challenging cases by over 30%. Importantly, alternative methods using electrons and ultraviolet light now achieved identification efficiency similar to the standard collision method, while delivering broader sequence coverage and more unique information about proteins.
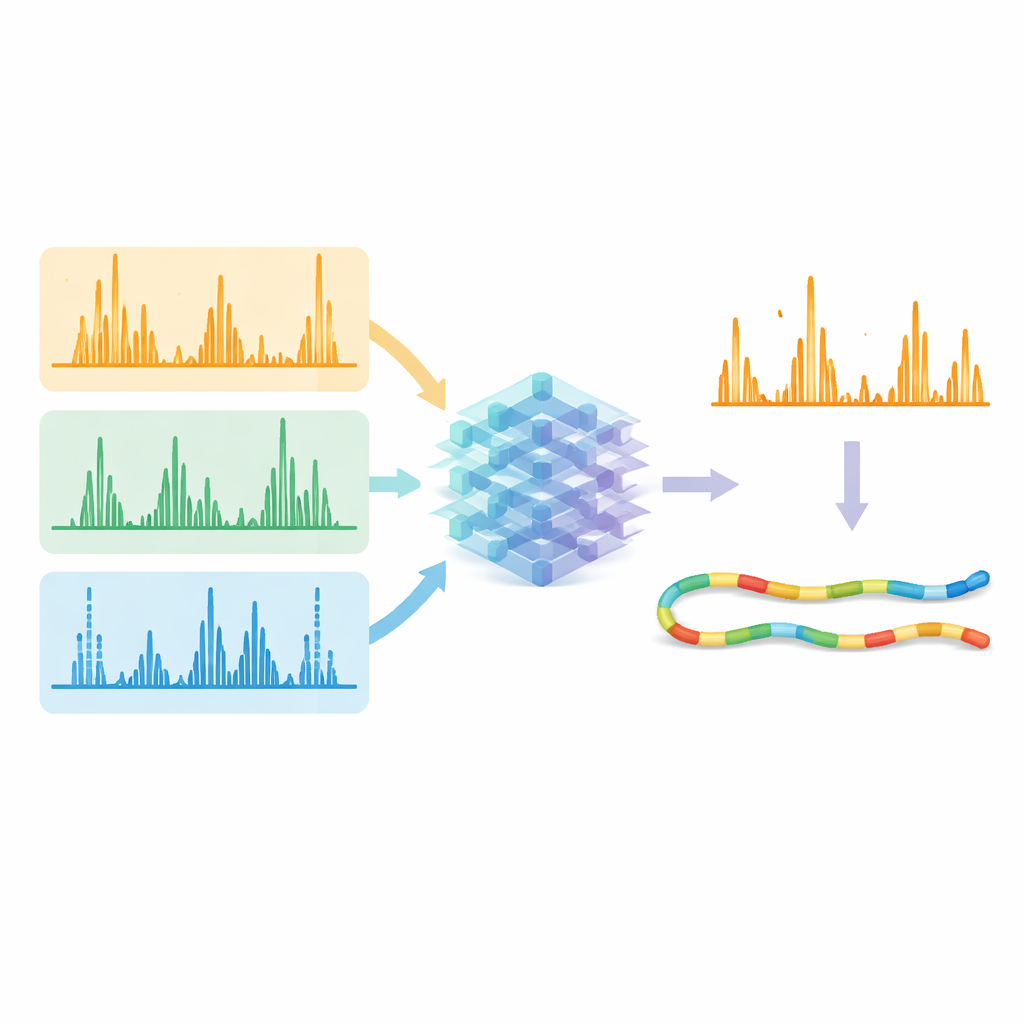
Bringing Advanced Methods into Everyday Use
Because the AI model is freely available and integrated into popular mass spectrometry software, it can be used not only for traditional, targeted measurements but also for newer data-independent acquisition strategies that scan wide swaths of the sample at once. Tests on human, plant and bacterial cell mixtures showed that the model generalizes well across species. In practical terms, this work removes a key barrier that had kept powerful but underused fragmentation methods confined to specialists. By unifying different ways of breaking proteins under one predictive model, the study provides a path to routine, high-coverage analysis of complex protein landscapes, making it easier for researchers to spot rare variants, map modifications and ultimately gain a more complete picture of how proteins behave in health and disease.
Citation: Levin, N., Saylan, C.C., Lapin, J. et al. Integration of alternative fragmentation techniques into standard LC-MS workflows using a single deep learning model enhances proteome coverage. Nat Methods 23, 805–814 (2026). https://doi.org/10.1038/s41592-026-03042-9
Keywords: proteomics, mass spectrometry, deep learning, protein fragmentation, spectral prediction