Clear Sky Science · sv
Integrering av alternativa fragmenteringstekniker i standard LC-MS-arbetsflöden med en enda djupinlärningsmodell förbättrar proteomtäckningen
Se mer av livets proteingear
Varje cell i din kropp innehåller tusentals olika proteiner, var och en med en specifik uppgift. Moderna masspektrometrimetoder kan redan läsa många av dessa proteiner genom att dela dem i bitar och väga fragmenten, men viktiga delar förblir osynliga—särskilt ovanliga proteinformer och subtila kemiska modifieringar som styr hälsa och sjukdom. Denna studie beskriver ett nytt sätt att kombinera flera avancerade fragmenteringsmetoder med en enda artificiell intelligensmodell så att forskare kan se mycket mer av proteinvärlden i ett rutinexperiment.
Hur proteiner vanligtvis läses
I de flesta laboratorier hakas proteiner först upp i mindre delar som kallas peptider och sedan förs in i ett instrument som separerar och väger dem. För att avgöra varje peptids sekvens krossar instrumentet medvetet dessa bitar och registrerar fragmentmönstret, ungefär som att krossa en vas och sluta sig till dess form från skärvorna. I åratal har en kollisionsbaserad metod—där peptider bryts genom stötar mot gaskulor—varit arbetshästen eftersom den är snabb, robust och väl understödd av mjukvara. Men detta standardförfarande har svårt att bevara känsliga kemiska märken på proteinet och missar delar av komplexa proteinformer, vilket lämnar blinda fläckar i vår biologiska förståelse.
Nya sätt att dela proteiner
Forskare har utvecklat andra sätt att knäcka peptider: med ultraviolett ljus eller elektronstrålar, som skär proteiner längs andra banor och ofta bevarar sköra egenskaper. Dessa angreppssätt kan generera rikare och mer informativa fragmentmönster, men de är långsammare, tekniskt krävande och dåligt stödja av analysverktyg. För att tackla detta byggde författarna vidare på ett specialiserat masspektrometer som kan tillämpa kollisions-, elektron- och fotonbaserade fragmenteringsmetoder i en och samma plattform och på den tidsskala som krävs för standardiserade vätskekromatografi–masspektrometri-arbetsflöden. De finjusterade driftsvillkoren för varje metod—såsom laserenergi eller elektronexponeringstid—så att varje metod gav så många användbara spektra som möjligt från komplexa mänskliga cellprover.
Bygga en enhetlig inlärningsmodell
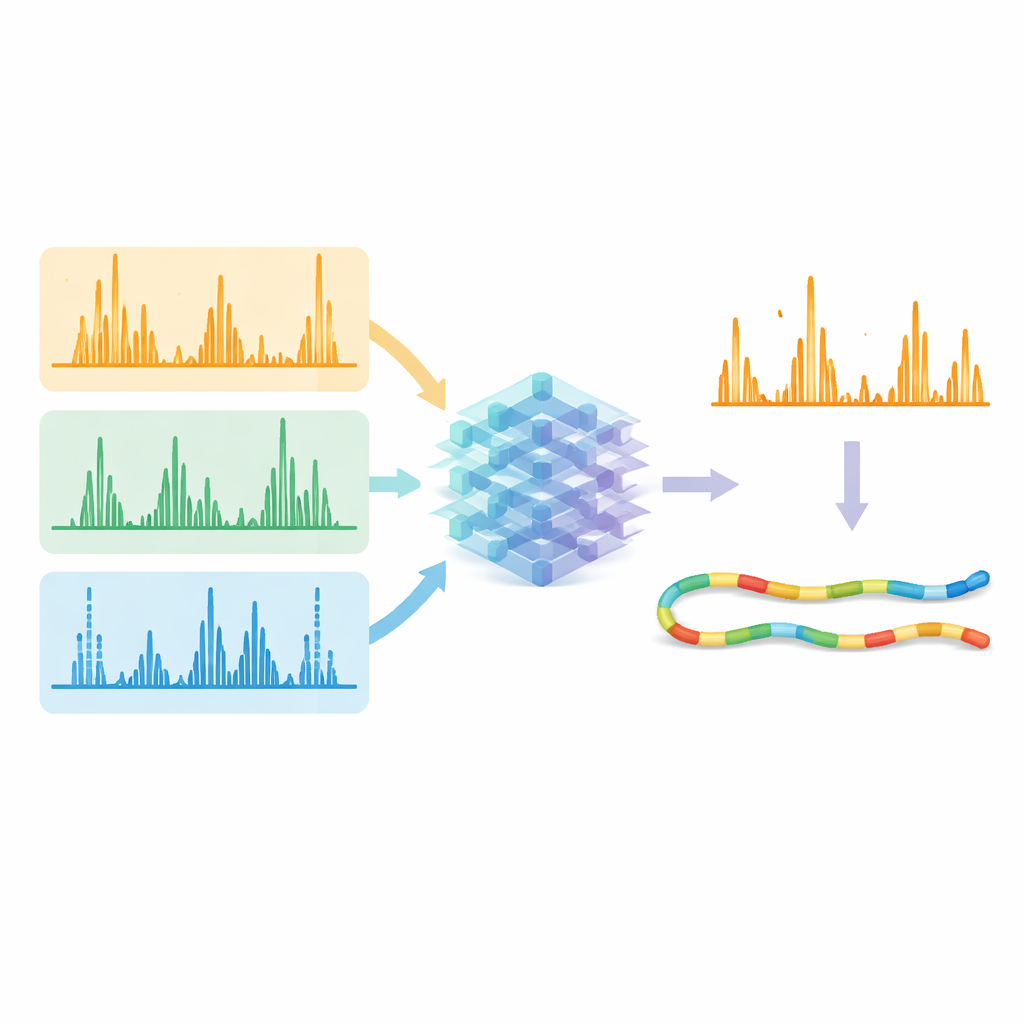
Med dessa optimerade metoder på plats genererade teamet omfattande dataset med hjälp av fem olika protein-snedknivar (enzymer), vilket gav en stor mångfald av peptidsekvenser. De använde sedan dessa dataset för att träna en enda djupinlärningsmodell, en förbättrad version av ett system kallat Prosit, för att förutsäga det detaljerade mönstret och intensiteten hos fragmenttoppar för alla fragmenteringstyper samtidigt. Istället för att behandla varje metod separat tar modellen som indata peptidsekvensen, dess laddning och vilken fragmenteringsmetod som användes, och ger som utdata de förväntade intensiteterna för hundratals möjliga fragmenttyper. De förutsagda spektrumen matchade experimentella data mycket väl över metoder, vilket visade att modellen effektivt hade lärt sig de karaktäristiska "fingeravtrycken" som genereras av ljus-, elektron- och kollisionsbaserad fragmentering.
Låta AI rensa upp signalen
Det verkliga testet var om dessa förutsägelser kunde förbättra hur många peptider som med hög säkerhet identifieras från rådata. Forskarna matade både de uppmätta spektrumen och de AI-förutsagda mönstren in i befintliga sök- och ompoängsättningsverktyg. När de bad mjukvaran fokusera på fragment som modellen sade borde vara starka och närvarande framträdde korrekta matchningar tydligare jämfört med felaktiga. Över data som samlats in med olika fragmenteringsmetoder och enzymer ökade antalet säkert identifierade peptid–spektrum-matchningar typiskt med mer än 10%, och i vissa utmanande fall med över 30%. Viktigt är att alternativa metoder med elektroner och ultraviolett ljus nu uppnådde identifieringseffektivitet liknande den standardiserade kollisionsmetoden, samtidigt som de levererade bredare sekvenstäckning och mer unik information om proteiner.
Göra avancerade metoder vardagliga
Då AI-modellen är fritt tillgänglig och integrerad i populär masspektrometrimjukvara kan den användas inte bara för traditionella, riktade mätningar utan också för nyare dataoberoende förvärvsstrategier som skannar stora delar av provet i taget. Tester på blandningar av mänskliga, växt- och bakterieceller visade att modellen generaliserar väl över arter. I praktiska termer tar detta arbete bort ett viktigt hinder som hållit kraftfulla men underutnyttjade fragmenteringsmetoder begränsade till specialister. Genom att förena olika sätt att bryta proteiner under en förutsägande modell ger studien en väg till rutinmässig, högtäckande analys av komplexa proteinlandskap, vilket gör det lättare för forskare att upptäcka sällsynta varianter, kartlägga modifieringar och i slutändan få en mer fullständig bild av hur proteiner beter sig i hälsa och sjukdom.
Citering: Levin, N., Saylan, C.C., Lapin, J. et al. Integration of alternative fragmentation techniques into standard LC-MS workflows using a single deep learning model enhances proteome coverage. Nat Methods 23, 805–814 (2026). https://doi.org/10.1038/s41592-026-03042-9
Nyckelord: proteomik, masspektrometri, djupinlärning, proteinnedbrytning, spektral prediktion