Clear Sky Science · es
Integración de técnicas alternativas de fragmentación en flujos de trabajo estándar de LC-MS mediante un único modelo de aprendizaje profundo mejora la cobertura del proteoma
Ver más de la maquinaria proteica de la vida
Cada célula de tu cuerpo contiene miles de proteínas distintas, cada una cumpliendo una tarea concreta. La espectrometría de masas moderna ya puede leer muchas de estas proteínas al romperlas en fragmentos y ponderar esos fragmentos, pero aún permanecen partes importantes invisibles, sobre todo formas proteicas inusuales y modificaciones químicas sutiles que determinan la salud y la enfermedad. Este estudio describe una nueva forma de combinar varias técnicas avanzadas de fragmentación con un único modelo de inteligencia artificial para que los científicos puedan ver mucho más del mundo proteico en un experimento de rutina.
Cómo se suelen leer las proteínas
En la mayoría de los laboratorios, las proteínas se trocean primero en piezas más pequeñas llamadas péptidos y luego se introducen en un instrumento que los separa y los pesa. Para determinar la secuencia de cada péptido, el aparato los rompe deliberadamente y registra el patrón de fragmentos, como destrozar un jarrón e inferir su forma a partir de los fragmentos. Durante años, un método basado en colisiones —donde los péptidos se fragmentan al chocar contra moléculas de gas— ha sido el caballo de batalla porque es rápido, robusto y está bien respaldado por software. Sin embargo, este enfoque estándar tiene dificultades para mantener intactas etiquetas químicas delicadas y pasa por alto partes de formas proteicas complejas, dejando puntos ciegos en nuestra comprensión de la biología.
Nuevas maneras de fragmentar proteínas
Los investigadores han desarrollado otras formas de abrir los péptidos: usando luz ultravioleta o haces de electrones, que desgarran las proteínas por rutas diferentes y con frecuencia preservan características frágiles. Estos enfoques pueden generar patrones de fragmentación más ricos e informativos, pero son más lentos, técnicamente exigentes y cuentan con escaso soporte en las herramientas de análisis de datos. Para afrontar esto, los autores partieron de un espectrómetro de masas especializado que puede aplicar métodos de ruptura basados en colisión, electrones y fotones en una sola plataforma y en la escala de tiempo necesaria para los flujos de trabajo estándar de cromatografía líquida–espectrometría de masas. Afinaron cuidadosamente las condiciones de funcionamiento de cada método —como la energía del láser o el tiempo de exposición a electrones— para que cada uno produjera la mayor cantidad posible de espectros útiles a partir de muestras celulares humanas complejas.
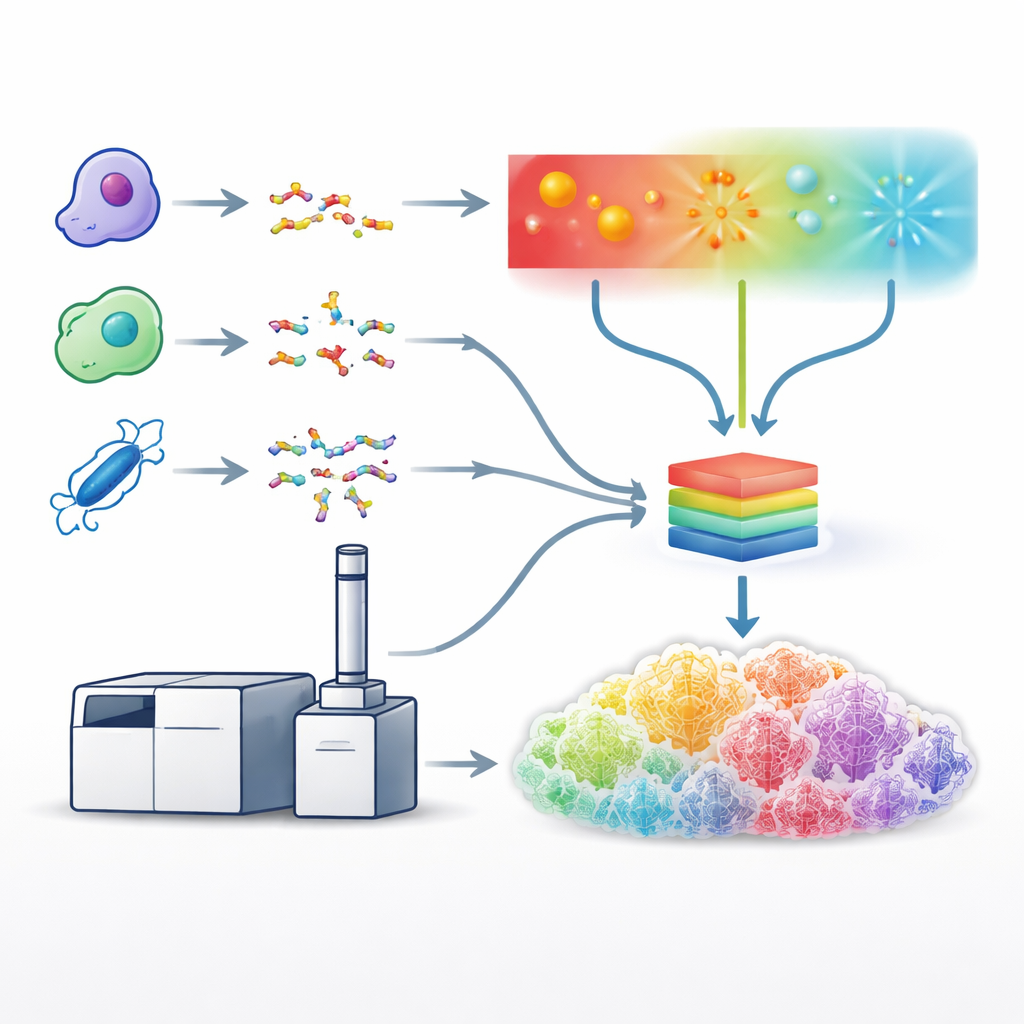
Construyendo un modelo de aprendizaje unificado
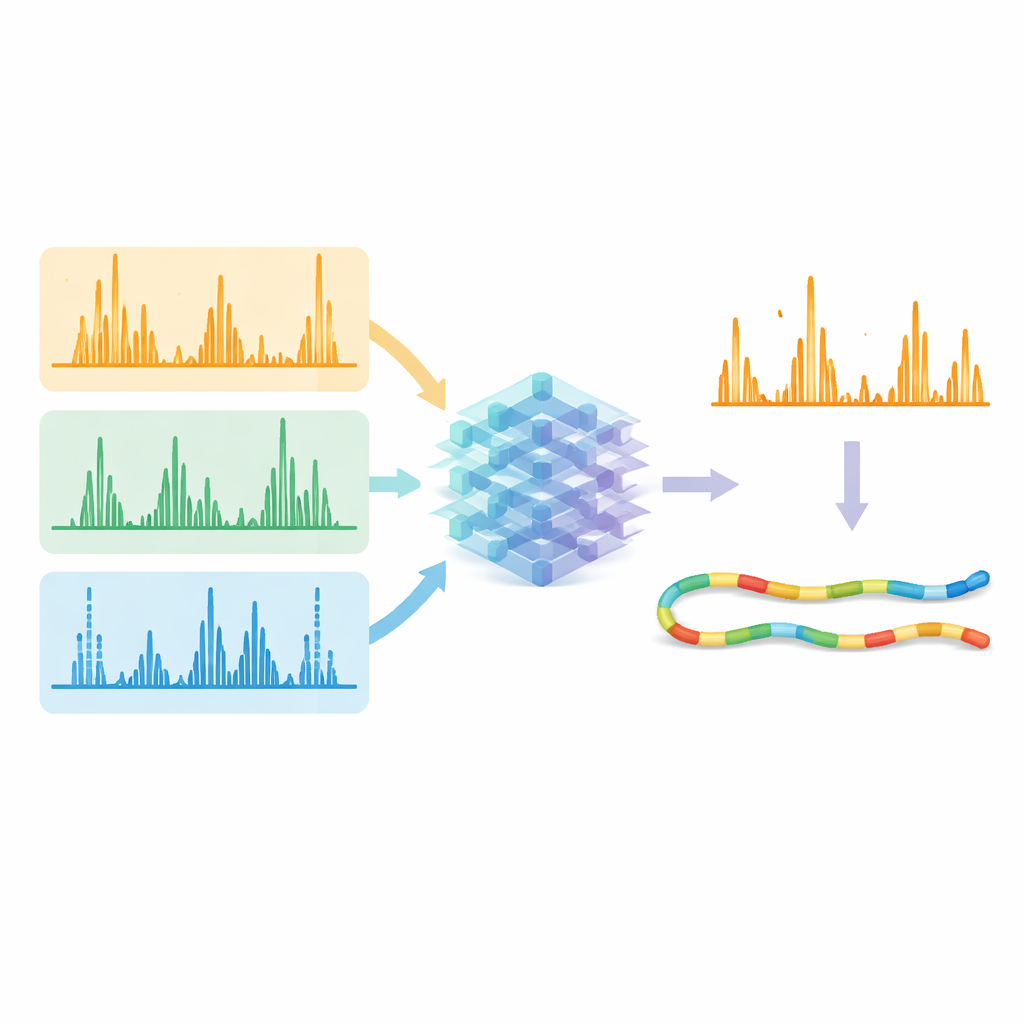
Con estos métodos optimizados en marcha, el equipo generó enormes conjuntos de datos usando cinco enzimas distintas para cortar proteínas, lo que produjo una gran diversidad de secuencias peptídicas. Luego usaron estos conjuntos para entrenar un único modelo de aprendizaje profundo, una versión mejorada de un sistema llamado Prosit, para predecir el patrón detallado y la intensidad de los picos de fragmentos para todos los tipos de fragmentación a la vez. En lugar de tratar cada método por separado, el modelo toma como entrada la secuencia del péptido, su carga y el método de fragmentación empleado, y proporciona las intensidades esperadas para cientos de tipos de fragmentos posibles. Los espectros predichos concordaron muy estrechamente con los datos experimentales a través de los métodos, lo que demuestra que el modelo había aprendido eficazmente las “huellas” características producidas por rupturas basadas en luz, electrones y colisiones.
Permitir que la IA limpie la señal
La prueba real fue si estas predicciones podían mejorar cuántos péptidos se identifican con confianza a partir de los datos crudos. Los investigadores introdujeron tanto los espectros medidos como los patrones predichos por la IA en las herramientas de búsqueda y revaloración existentes. Cuando pidieron al software que se enfocara en los fragmentos que el modelo indicaba que deberían ser fuertes y estar presentes, las coincidencias correctas destacaron con mayor claridad frente a las falsas. En los datos recogidos con distintos métodos de fragmentación y enzimas, el número de correspondencias péptido–espectro identificadas con confianza aumentó típicamente más de un 10 %, y en algunos casos desafiantes superó el 30 %. Es importante que los métodos alternativos que usan electrones y luz ultravioleta alcanzaran ahora una eficiencia de identificación similar a la del método estándar de colisiones, a la vez que ofrecían una cobertura de secuencia más amplia y más información única sobre las proteínas.
Incorporar métodos avanzados al uso cotidiano
Como el modelo de IA está disponible de forma gratuita e integrado en software popular de espectrometría de masas, puede emplearse no solo para mediciones tradicionales y dirigidas sino también para estrategias más recientes de adquisición independiente de datos que escanean amplias porciones de la muestra a la vez. Pruebas en mezclas de células humanas, vegetales y bacterianas mostraron que el modelo se generaliza bien entre especies. En términos prácticos, este trabajo elimina una barrera clave que había mantenido a métodos de fragmentación poderosos pero subutilizados confinados a especialistas. Al unificar diferentes formas de fragmentar proteínas bajo un modelo predictivo, el estudio ofrece un camino hacia un análisis de rutina y de alta cobertura de paisajes proteicos complejos, facilitando a los investigadores detectar variantes raras, cartografiar modificaciones y, en última instancia, obtener una imagen más completa de cómo se comportan las proteínas en la salud y la enfermedad.
Cita: Levin, N., Saylan, C.C., Lapin, J. et al. Integration of alternative fragmentation techniques into standard LC-MS workflows using a single deep learning model enhances proteome coverage. Nat Methods 23, 805–814 (2026). https://doi.org/10.1038/s41592-026-03042-9
Palabras clave: proteómica, espectrometría de masas, aprendizaje profundo, fragmentación de proteínas, predicción espectral