Clear Sky Science · ja
単一の深層学習モデルで複数の断片化手法を標準的なLC–MSワークフローに統合するとプロテオーム被覆が向上する
生命のタンパク質機構をより広く捉える
体内のあらゆる細胞には、特定の役割を担う何千もの異なるタンパク質が詰まっています。現代の質量分析はこれらのタンパク質の多くを、タンパク質を断片化して得られる断片の質量を測ることで読み取ることができますが、重要な部分は依然として見えにくいままです。特に、変わった形のタンパク質や健康と病気に影響する微細な化学的修飾は検出が難しいことが多い。本研究は、いくつかの高度な断片化手法を単一の人工知能モデルで組み合わせる新しい方法を示しており、通常の実験で見ることのできるタンパク質の世界を大幅に広げる可能性を示しています。
タンパク質の一般的な読み取り方
多くの研究室では、まずタンパク質をペプチドと呼ばれる小さな断片に切断し、これを分離して質量を測定する装置にかけます。各ペプチドの配列を決定するために、装置は意図的にこれらの断片を破砕し、破片のパターンを記録します。これは壺を粉々にして破片から元の形を推測するようなものです。長年、ペプチドを気体分子と衝突させて破壊する衝突型の手法が主力として使われてきました。これは高速で堅牢、かつソフトウェアのサポートが充実しているためです。しかし、この標準的手法は繊細な化学的タグを保持するのが苦手で、複雑なタンパク質形態の一部を見逃し、生物学の理解に盲点を残してしまいます。
タンパク質を切り分ける新しい方法
研究者たちは、紫外線や電子ビームなど、ペプチドを別の経路で切断する手法も開発してきました。これらは壊れる経路が異なり、壊れやすい特徴を保持しやすいことが多く、より豊かで情報量の高い断片パターンを生み出すことがあります。しかし、これらの手法は遅く、技術的に要求が高く、データ解析ツールのサポートも乏しいという欠点があります。これに対処するため、著者らは衝突、電子、光(光子)に基づく断片化を一つのプラットフォーム上で、標準的な液体クロマトグラフィ—質量分析(LC–MS)の時間スケールで適用できる特殊な質量分析計を基に構築しました。各手法についてレーザーエネルギーや電子照射時間など動作条件を慎重に調整し、複雑なヒト細胞サンプルからできるだけ多くの有用なスペクトルを得られるようにしました。
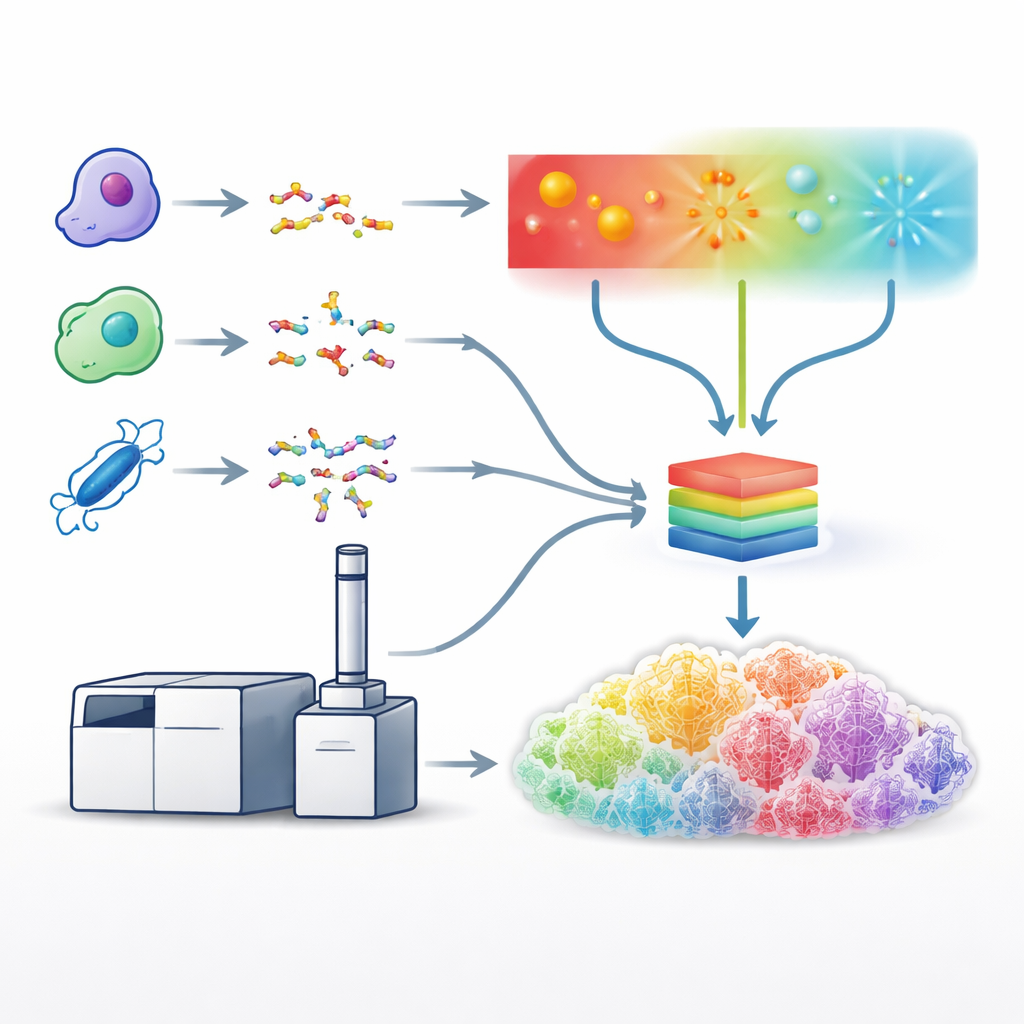
統一的な学習モデルの構築
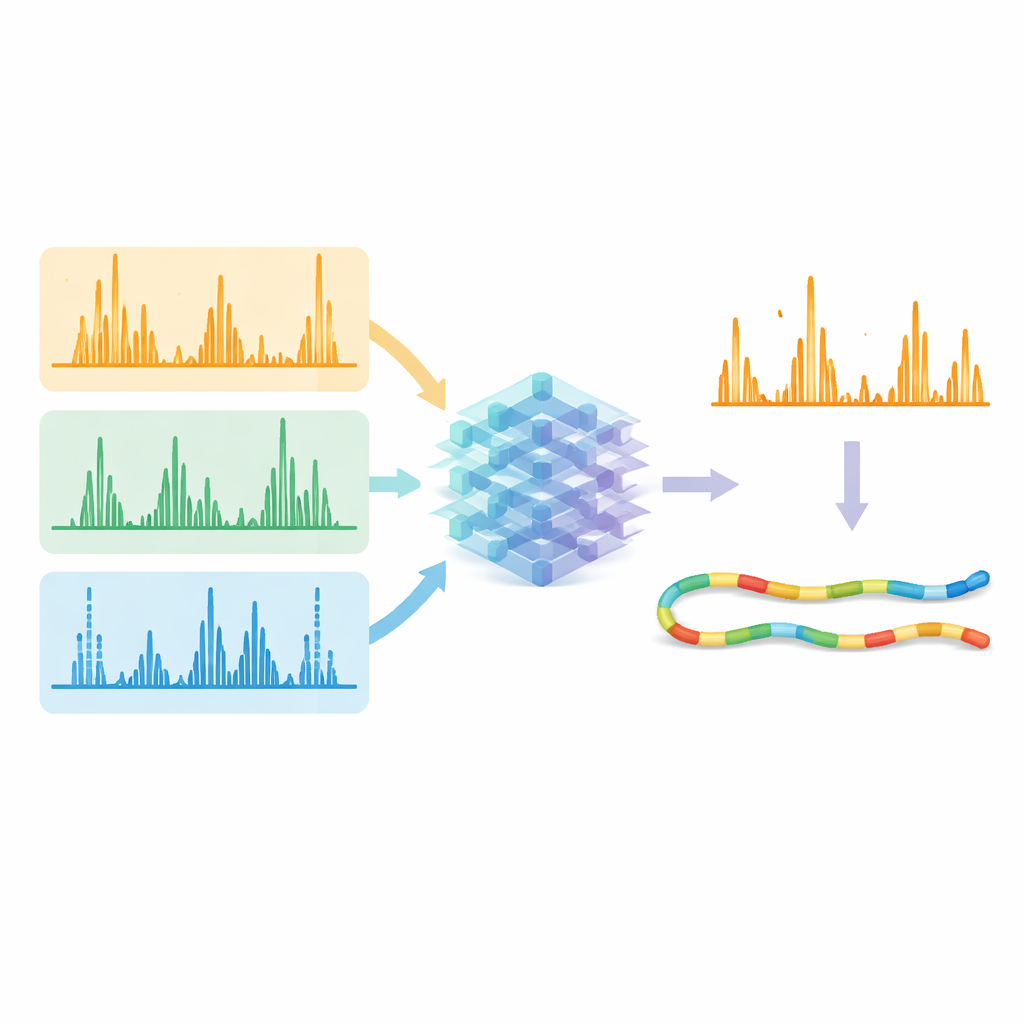
これらの最適化された手法を用いて、チームは5種類のタンパク質切断酵素を使った膨大なデータセットを生成し、多様なペプチド配列を網羅しました。これらのデータを使って、Prositというシステムを拡張した単一の深層学習モデルを訓練し、すべての断片化タイプに対して断片ピークの詳細なパターンと強度を同時に予測できるようにしました。各手法を個別に扱う代わりに、モデルは入力としてペプチド配列、荷電状態、使用された断片化手法を受け取り、数百に及ぶ可能な断片タイプそれぞれの期待強度を出力します。予測されたスペクトルは実験データと非常に高い一致を示し、光、電子、衝突に基づく断片化が作り出す特徴的な“フィンガープリント”をモデルが効果的に学習したことを示しました。
AIで信号を磨く
真の検証は、これらの予測が生データから確実に同定されるペプチド数を増やせるかどうかでした。研究者たちは測定されたスペクトルとAIが予測したパターンの両方を既存の検索および再スコアリングツールに入力しました。ソフトウェアに対してモデルが「強く出るはずだ」と示した断片に注目させると、正しい一致が誤りのものよりも際立つようになりました。異なる断片化手法や酵素で収集されたデータを通して、確信度の高いペプチド—スペクトル一致の数は通常10%以上増加し、難しいケースでは30%を超える増加が見られました。重要なのは、電子や紫外光を用いる代替手法も、これまでの衝突法と同等の同定効率を達成しつつ、配列被覆を広げ、タンパク質に関するより独自の情報を提供できるようになった点です。
高度な手法を日常に取り込む
このAIモデルは無償で利用可能であり、一般的な質量分析ソフトウェアに統合されているため、従来のターゲット測定だけでなく、サンプルの広範囲を同時にスキャンするデータ非依存取得(DIA)などの新しい戦略にも使えます。ヒト、植物、細菌の混合サンプルでのテストは、モデルが種を越えてよく一般化することを示しました。実践的には、この研究は強力だがこれまで専門家に限られていた断片化手法の利用障壁を取り除きます。異なる断片化手法を一つの予測モデルで統一することで、複雑なタンパク質景観のルーチンかつ高被覆の解析への道筋を示し、希少変異の検出、修飾のマッピング、そして最終的には健康と病気におけるタンパク質の振る舞いをより完全に把握することを容易にします。
引用: Levin, N., Saylan, C.C., Lapin, J. et al. Integration of alternative fragmentation techniques into standard LC-MS workflows using a single deep learning model enhances proteome coverage. Nat Methods 23, 805–814 (2026). https://doi.org/10.1038/s41592-026-03042-9
キーワード: プロテオミクス, 質量分析法, 深層学習, タンパク質断片化, スペクトル予測