Clear Sky Science · nl
Integratie van alternatieve fragmentatietechnieken in standaard LC-MS-workflows met een enkel deep learning-model vergroot de proteoomdekking
Meer zien van het eiwitmachinaërie van het leven
Elke cel in je lichaam bevat duizenden verschillende eiwitten, elk met een specifieke taak. Moderne massaspectrometrie kan veel van deze eiwitten al detecteren door ze in stukjes te breken en de fragmenten te wegen, maar belangrijke onderdelen blijven onzichtbaar—vooral ongebruikelijke eiwitvormen en subtiele chemische aanpassingen die gezondheid en ziekte aansturen. Deze studie beschrijft een nieuwe manier om verschillende geavanceerde fragmentatiemethoden te combineren met één enkel kunstmatig-intelligentie-model, zodat wetenschappers veel meer van de eiwitwereld kunnen zien in een routinematig experiment.
Hoe eiwitten gewoonlijk worden gelezen
In de meeste laboratoria worden eiwitten eerst geknipt in kleinere stukken, peptiden genoemd, en vervolgens in een instrument gebracht dat ze scheidt en weegt. Om de volgorde van elk peptide te bepalen, laat het instrument deze stukjes opzettelijk uiteenspatten en registreert het patroon van fragmenten—alsof je een vaas kapot slaat en uit de scherven de vorm probeert af te leiden. Jarenlang was een botsingsgebaseerde methode—waarbij peptiden breken door te botsen met gasmoleculen—de standaard, omdat deze snel, robuust en goed ondersteund door software is. Deze standaardaanpak heeft echter moeite om kwetsbare chemische labels op het eiwit intact te houden en mist delen van complexe eiwitvormen, waardoor er blinde vlekken in ons begrip van biologie ontstaan.
Nieuwe manieren om eiwitten te fragmenteren
Onderzoekers hebben andere manieren ontwikkeld om peptiden te splijten: met ultraviolet licht of met elektronenbundels, die eiwitten langs andere paden doorsnijden en vaak fragiele kenmerken behouden. Deze benaderingen kunnen rijkere en informatieverrerende fragmentpatronen opleveren, maar ze zijn trager, technisch veeleisender en slecht ondersteund door data-analysetools. Om dit aan te pakken bouwden de auteurs voort op een gespecialiseerd massaspectrometer dat botsings-, elektron- en fotongebaseerde fragmentatiemethoden op één platform kan toepassen en op de tijdschaal die nodig is voor standaard vloeistofchromatografie–massaspectrometrie-workflows. Ze stemden de bedrijfscondities voor elke methode zorgvuldig af—zoals laserenergie of elektronblootstellingstijd—zodat elk zoveel mogelijk bruikbare spectra uit complexe menselijke celmonsters genereerde.
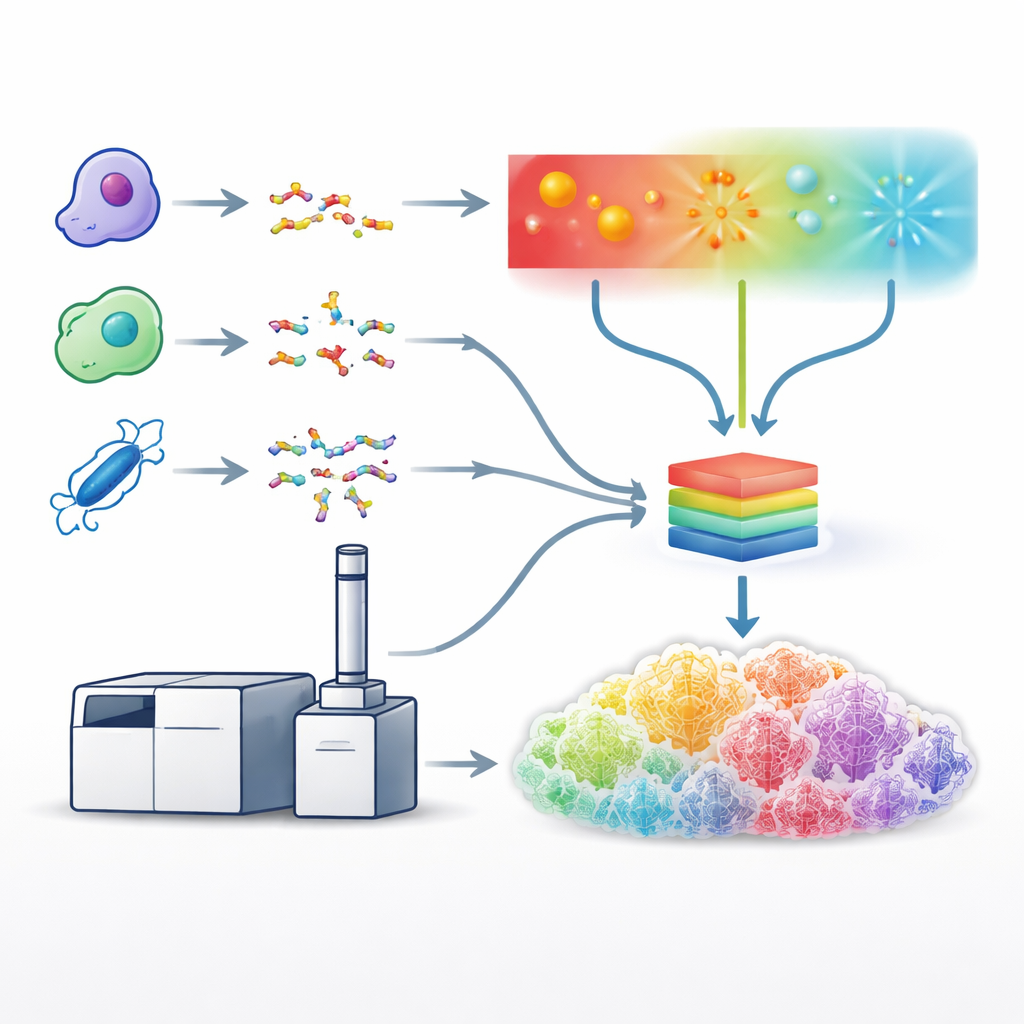
Het bouwen van een verenigd leer-model
Met deze geoptimaliseerde methoden produceerde het team enorme datasets met vijf verschillende eiwitknipende enzymen, wat zorgde voor een grote diversiteit aan peptidesequenties. Ze gebruikten deze datasets om één enkel deep learning-model te trainen, een verbeterde versie van een systeem genaamd Prosit, om het gedetailleerde patroon en de intensiteit van fragmentpieken voor alle fragmentatietypen tegelijk te voorspellen. In plaats van elke methode afzonderlijk te behandelen, neemt het model als invoer de peptidesequentie, de lading en welke fragmentatiemethode werd gebruikt, en geeft het de verwachte intensiteiten voor honderden mogelijke fragmenttypen als uitvoer. De voorspelde spectra kwamen zeer nauwkeurig overeen met experimentele gegevens over de verschillende methoden heen, wat liet zien dat het model effectief de karakteristieke ‘vingerafdrukken’ had geleerd die door licht-, elektron- en botsingsgebaseerde fragmentatie worden geproduceerd.
AI laten het signaal opschonen
De echte test was of deze voorspellingen konden verbeteren hoeveel peptiden met vertrouwen uit de ruwe data worden geïdentificeerd. De onderzoekers voerden zowel de gemeten spectra als de door AI voorspelde patronen in bestaande zoek- en herwaarderingstools. Toen ze de software vroegen zich te concentreren op fragmenten die het model als sterk en aanwezig voorspelde, kwamen correcte overeenkomsten duidelijker naar voren ten opzichte van foutieve. Over datasets verzameld met verschillende fragmentatiemethoden en enzymen steeg het aantal met vertrouwen geïdentificeerde peptide–spectrum-overeenkomsten doorgaans met meer dan 10%, en in sommige uitdagende gevallen met meer dan 30%. Belangrijk is dat alternatieve methoden met elektronen en ultraviolet licht nu een identificefficiëntie bereikten vergelijkbaar met de standaard botsingsmethode, terwijl ze een bredere sequentiedekking en meer unieke informatie over eiwitten leverden.
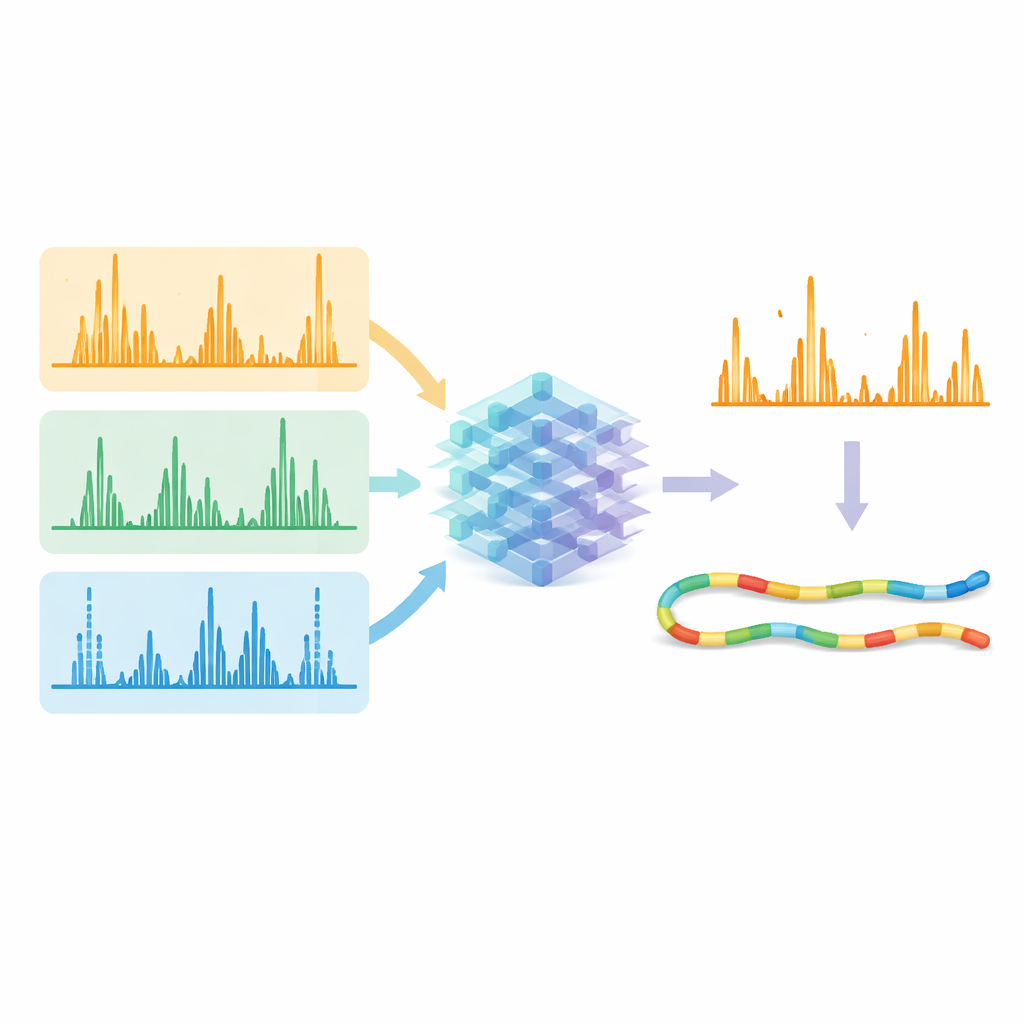
Geavanceerde methoden alledaags maken
Aangezien het AI-model vrij beschikbaar is en is geïntegreerd in populaire massaspectrometriesoftware, kan het niet alleen worden gebruikt voor traditionele, gerichte metingen maar ook voor nieuwere data-onafhankelijke acquisitiestrategieën die brede delen van het monster tegelijk scannen. Tests op menselijke, plantaardige en bacteriële celmengsels toonden aan dat het model goed generaliseert over soorten heen. In praktische termen verwijdert dit werk een belangrijke barrière die krachtige maar weinig gebruikte fragmentatiemethoden tot nu toe aan specialisten bond. Door verschillende manieren om eiwitten te fragmenteren onder één voorspellend model te verenigen, biedt de studie een pad naar routinematige, hoogdekkinganalyse van complexe eiwitlandschappen, waardoor het voor onderzoekers makkelijker wordt zeldzame varianten te herkennen, modificaties in kaart te brengen en uiteindelijk een vollediger beeld te krijgen van hoe eiwitten zich gedragen in gezondheid en ziekte.
Bronvermelding: Levin, N., Saylan, C.C., Lapin, J. et al. Integration of alternative fragmentation techniques into standard LC-MS workflows using a single deep learning model enhances proteome coverage. Nat Methods 23, 805–814 (2026). https://doi.org/10.1038/s41592-026-03042-9
Trefwoorden: proteomica, massaspectrometrie, deep learning, eiwitfragmentatie, spectrumvoorspelling