Clear Sky Science · pl
Integracja alternatywnych technik fragmentacji ze standardowymi przepływami LC-MS przy użyciu jednego modelu głębokiego uczenia zwiększa pokrycie proteomu
Widzieć więcej maszynerii białkowej życia
Każda komórka w twoim ciele zawiera tysiące różnych białek, z których każde pełni określoną funkcję. Współczesna spektrometria mas potrafi już odczytać wiele z tych białek poprzez rozbicie ich na fragmenty i zważenie fragmentów, lecz istotne części wciąż pozostają niewidoczne — szczególnie nietypowe formy białek oraz subtelne modyfikacje chemiczne, które wpływają na zdrowie i choroby. W tym badaniu opisano nowy sposób łączenia kilku zaawansowanych metod fragmentacji z jednym modelem sztucznej inteligencji, dzięki czemu naukowcy mogą zobaczyć znacznie więcej świata białek podczas rutynowego eksperymentu.
Jak zwykle odczytuje się białka
W większości laboratoriów białka najpierw są rozdrabniane na krótsze fragmenty zwane peptydami, a następnie wprowadzane do urządzenia, które je separuje i waży. Aby ustalić sekwencję peptydu, instrument celowo rozbija te fragmenty i rejestruje wzór powstałych jonów fragmentacyjnych — jak rozbicie wazonu i wywnioskowanie jego kształtu na podstawie odłamków. Przez lata metodą dominującą była fragmentacja zderzeniowa — peptydy są rozrywane przez zderzenia z cząsteczkami gazu — ponieważ jest szybka, niezawodna i dobrze obsługiwana przez oprogramowanie. Jednak to standardowe podejście ma problemy z zachowaniem delikatnych znaczników chemicznych na białkach i pomija elementy złożonych form białkowych, pozostawiając luki w naszym zrozumieniu biologii.
Nowe sposoby łamania białek
Naukowcy opracowali inne metody rozcinania peptydów: użycie ultrafioletowego światła lub wiązek elektronów, które przecinają białka innymi drogami i często zachowują kruche cechy. Podejścia te potrafią generować bogatsze i bardziej informatywne wzory fragmentów, ale są wolniejsze, wymagają zaawansowanej obsługi i słabo wspierane przez narzędzia analizy danych. Aby temu sprostać, autorzy oparli się na wyspecjalizowanym spektrometrze mas zdolnym do stosowania metod zderzeniowych, elektronowych i fotonowych na jednej platformie i w czasie kompatybilnym ze standardowymi przepływami chromatografii cieczowej–spektrometrii mas. Precyzyjnie dostroili warunki pracy dla każdej metody — na przykład energię lasera czy czas ekspozycji elektronów — tak, by każda dawała jak najwięcej użytecznych widm z złożonych próbek ludzkich komórek.
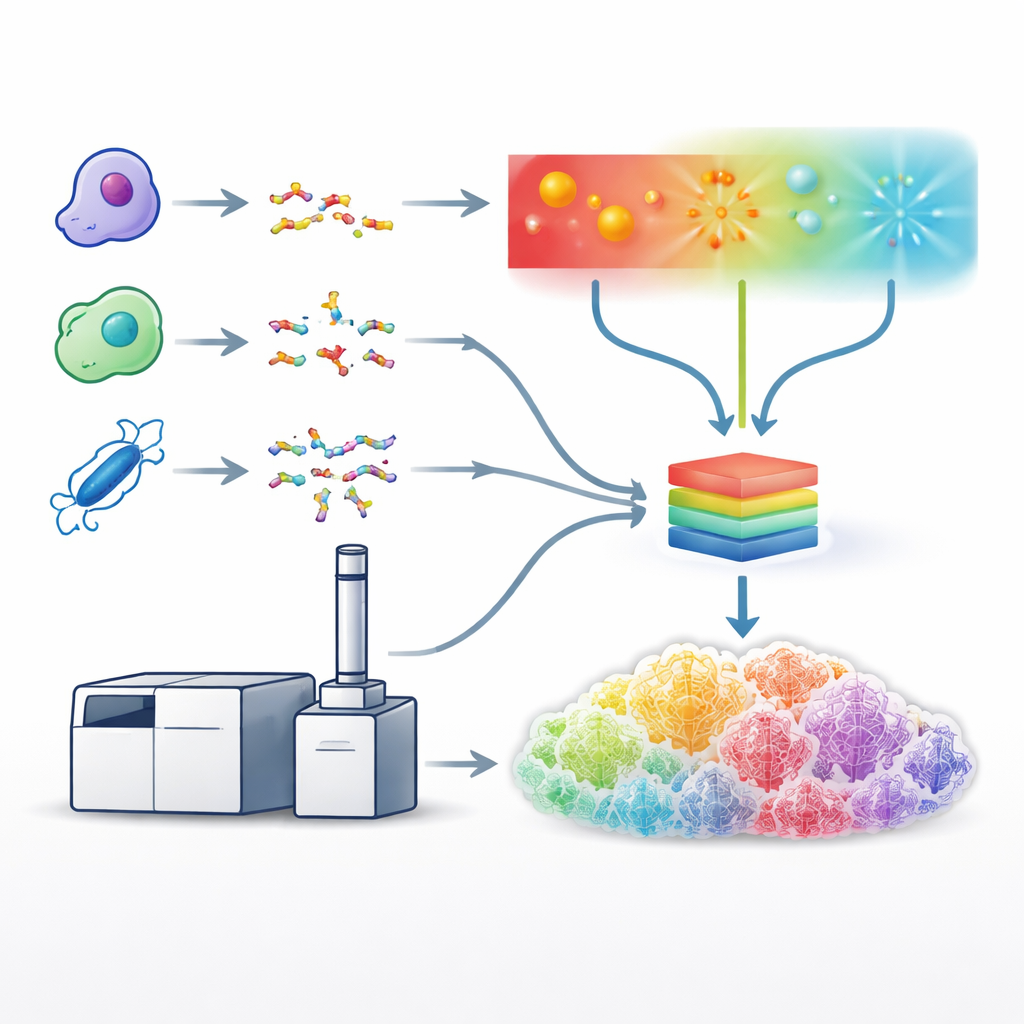
Budowa zunifikowanego modelu uczącego się
Z tymi zoptymalizowanymi metodami zespół wygenerował obszerne zbiory danych wykorzystując pięć różnych enzymów tnących białka, co dało ogromną różnorodność sekwencji peptydów. Następnie użyli tych danych do wytrenowania jednego modelu głębokiego uczenia — ulepszonej wersji systemu o nazwie Prosit — aby jednocześnie przewidywał szczegółowy wzór i intensywność pików fragmentacyjnych dla wszystkich typów fragmentacji. Zamiast traktować każdą metodę osobno, model przyjmuje jako wejście sekwencję peptydu, jego ładunek oraz zastosowaną metodę fragmentacji, a na wyjściu podaje oczekiwane intensywności setek możliwych typów fragmentów. Przewidywane widma bardzo dobrze odpowiadały danym eksperymentalnym we wszystkich metodach, co pokazało, że model skutecznie nauczył się charakterystycznych „odcisków palców” generowanych przez łamanie światłem, elektronami i zderzeniami.
Pozwolenie AI na oczyszczenie sygnału
Prawdziwe sprawdzenie polegało na tym, czy te przewidywania mogą poprawić liczbę peptydów, które z surowych danych identyfikuje się z pewnością. Badacze wprowadzili zarówno zmierzone widma, jak i wzory przewidziane przez AI do istniejących narzędzi wyszukiwania i ponownego oceniania wyników. Gdy poprosili oprogramowanie, aby skupiło się na fragmentach, które model wskazywał jako silne i obecne, prawidłowe dopasowania wyraźniej odróżniały się od fałszywych. W danych zebranych przy różnych metodach fragmentacji i przy użyciu różnych enzymów liczba pewnych dopasowań peptyd–widmo zazwyczaj wzrosła o ponad 10%, a w niektórych trudnych przypadkach o ponad 30%. Co ważne, alternatywne metody wykorzystujące elektrony i ultrafiolet osiągnęły teraz efektywność identyfikacji zbliżoną do standardowej metody zderzeniowej, jednocześnie zapewniając szersze pokrycie sekwencji i więcej unikatowych informacji o białkach.
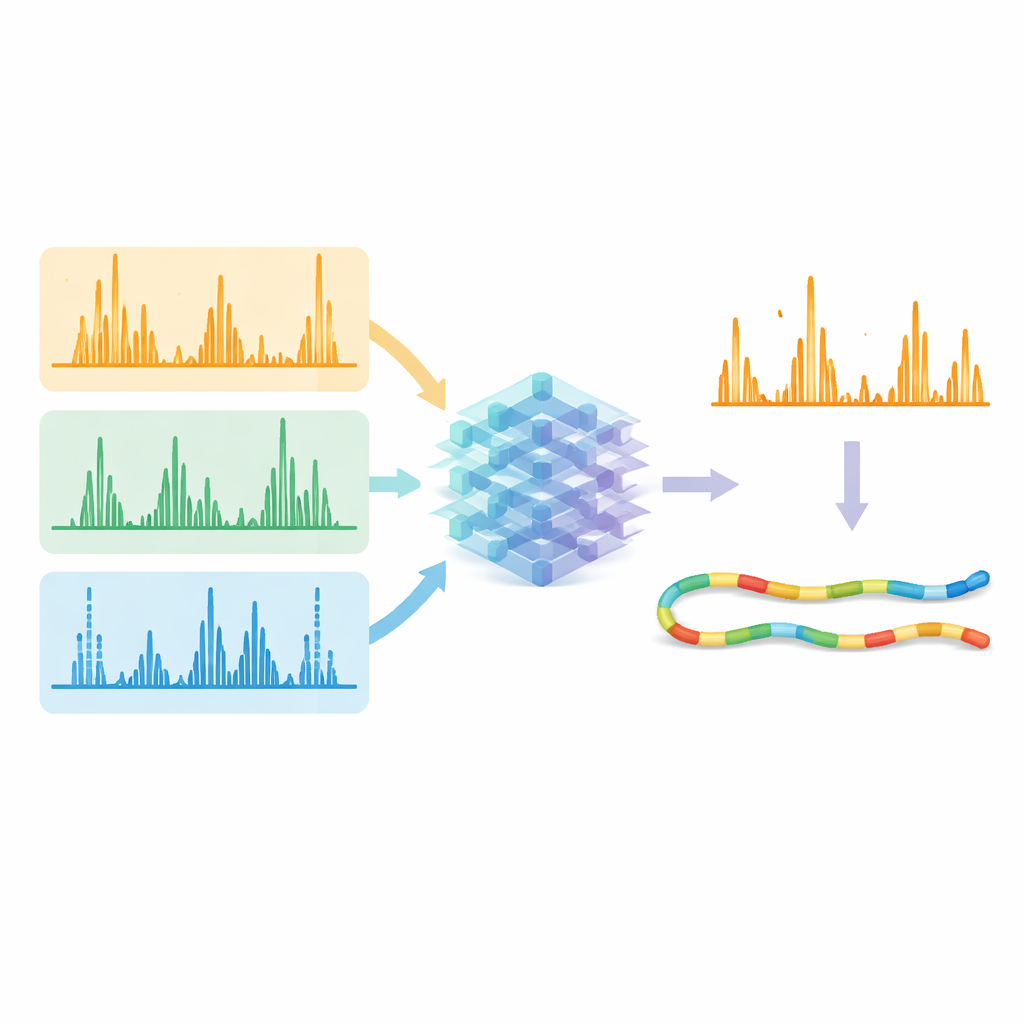
Wprowadzenie zaawansowanych metod do codziennego użytku
Ponieważ model AI jest udostępniony bezpłatnie i zintegrowany z popularnym oprogramowaniem do spektrometrii mas, można go używać nie tylko do tradycyjnych, celowanych pomiarów, lecz także do nowszych strategii akwizycji niezależnej od danych (data-independent acquisition), które skanują duże fragmenty próbki na raz. Testy przeprowadzone na mieszankach komórek ludzkich, roślinnych i bakteryjnych wykazały, że model dobrze uogólnia się między gatunkami. W praktyce praca ta usuwa kluczową barierę, która dotąd ograniczała potężne, lecz rzadko używane metody fragmentacji do kręgu specjalistów. Zjednoczenie różnych sposobów rozbijania białek pod jednym modelem predykcyjnym otwiera drogę do rutynowej, szerokopokryciowej analizy złożonych krajobrazów białkowych, ułatwiając badaczom wykrywanie rzadkich wariantów, mapowanie modyfikacji i ostatecznie uzyskanie pełniejszego obrazu zachowania białek w zdrowiu i chorobie.
Cytowanie: Levin, N., Saylan, C.C., Lapin, J. et al. Integration of alternative fragmentation techniques into standard LC-MS workflows using a single deep learning model enhances proteome coverage. Nat Methods 23, 805–814 (2026). https://doi.org/10.1038/s41592-026-03042-9
Słowa kluczowe: proteomika, spektrometria mas, głębokie uczenie, fragmentacja białek, predykcja widm