Clear Sky Science · it
Integrazione di tecniche alternative di frammentazione nei flussi di lavoro LC-MS standard tramite un unico modello di deep learning migliora la copertura del proteoma
Vedere di più della macchina proteica della vita
Ogni cellula del tuo corpo è piena di migliaia di proteine diverse, ognuna con un compito specifico. La spettrometria di massa moderna riesce già a leggere molte di queste proteine spezzandole in frammenti e pesando i frammenti, ma parti importanti restano invisibili—soprattutto forme proteiche insolite e modifiche chimiche sottili che guidano salute e malattia. Questo studio descrive un nuovo modo di combinare diverse tecniche avanzate di frammentazione con un unico modello di intelligenza artificiale in modo che gli scienziati possano vedere molto di più del mondo proteico in un esperimento di routine.
Come vengono di solito lette le proteine
Nella maggior parte dei laboratori, le proteine vengono prima tagliate in pezzi più piccoli chiamati peptidi e poi introdotte in uno strumento che li separa e li pesa. Per determinare la sequenza di ciascun peptide, lo strumento lo frantuma deliberatamente e registra il modello dei frammenti, come rompere un vaso e dedurne la forma dai cocci. Per anni, un metodo basato sulle collisioni—dove i peptidi vengono rotti urtando molecole di gas—è stato il cavallo di battaglia perché è veloce, robusto e ben supportato dal software. Tuttavia, questo approccio standard fatica a mantenere intatti i marcatori chimici delicati e perde parti delle forme proteiche complesse, lasciando punti ciechi nella nostra comprensione della biologia.
Nuovi modi per frammentare le proteine
I ricercatori hanno sviluppato altri modi per spaccare i peptidi: usando luce ultravioletta o fasci di elettroni, che tagliano le proteine lungo percorsi diversi e spesso preservano caratteristiche fragili. Questi approcci possono generare schemi di frammentazione più ricchi e informativi, ma sono più lenti, tecnicamente esigenti e scarsamente supportati dagli strumenti di analisi dei dati. Per affrontare questo problema, gli autori si sono basati su uno spettrometro di massa specializzato in grado di applicare metodi di frammentazione basati su collisioni, elettroni e fotoni su un’unica piattaforma e nelle tempistiche richieste dai flussi di lavoro standard di cromatografia liquida–spettrometria di massa. Hanno messo a punto con cura le condizioni operative per ciascun metodo—come l’energia del laser o il tempo di esposizione agli elettroni—così che ciascuno producesse il maggior numero possibile di spettri utili da campioni complessi di cellule umane.
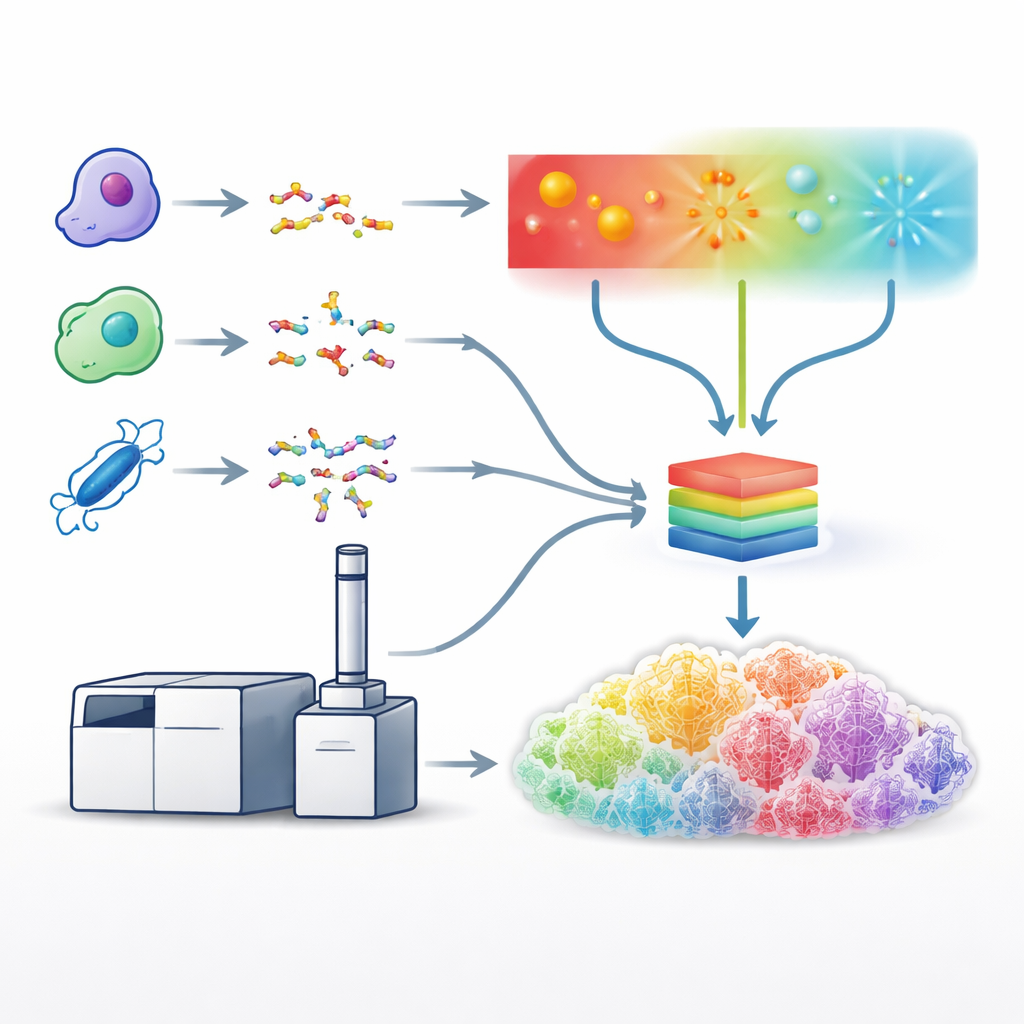
Costruire un modello di apprendimento unificato
Con questi metodi ottimizzati, il team ha generato dataset vastissimi usando cinque diversi enzimi di taglio proteico, che hanno fornito una grande diversità di sequenze peptidiche. Hanno poi usato questi dataset per addestrare un unico modello di deep learning, una versione migliorata di un sistema chiamato Prosit, per predire il pattern dettagliato e l’intensità dei picchi di frammento per tutti i tipi di frammentazione contemporaneamente. Invece di trattare ogni metodo separatamente, il modello prende in input la sequenza del peptide, la sua carica e il metodo di frammentazione usato, e restituisce le intensità attese per centinaia di possibili tipi di frammento. Gli spettri predetti corrispondevano molto da vicino ai dati sperimentali attraverso i metodi, dimostrando che il modello aveva effettivamente appreso le «impronte» caratteristiche prodotte dalla frammentazione basata su luce, elettroni e collisioni.
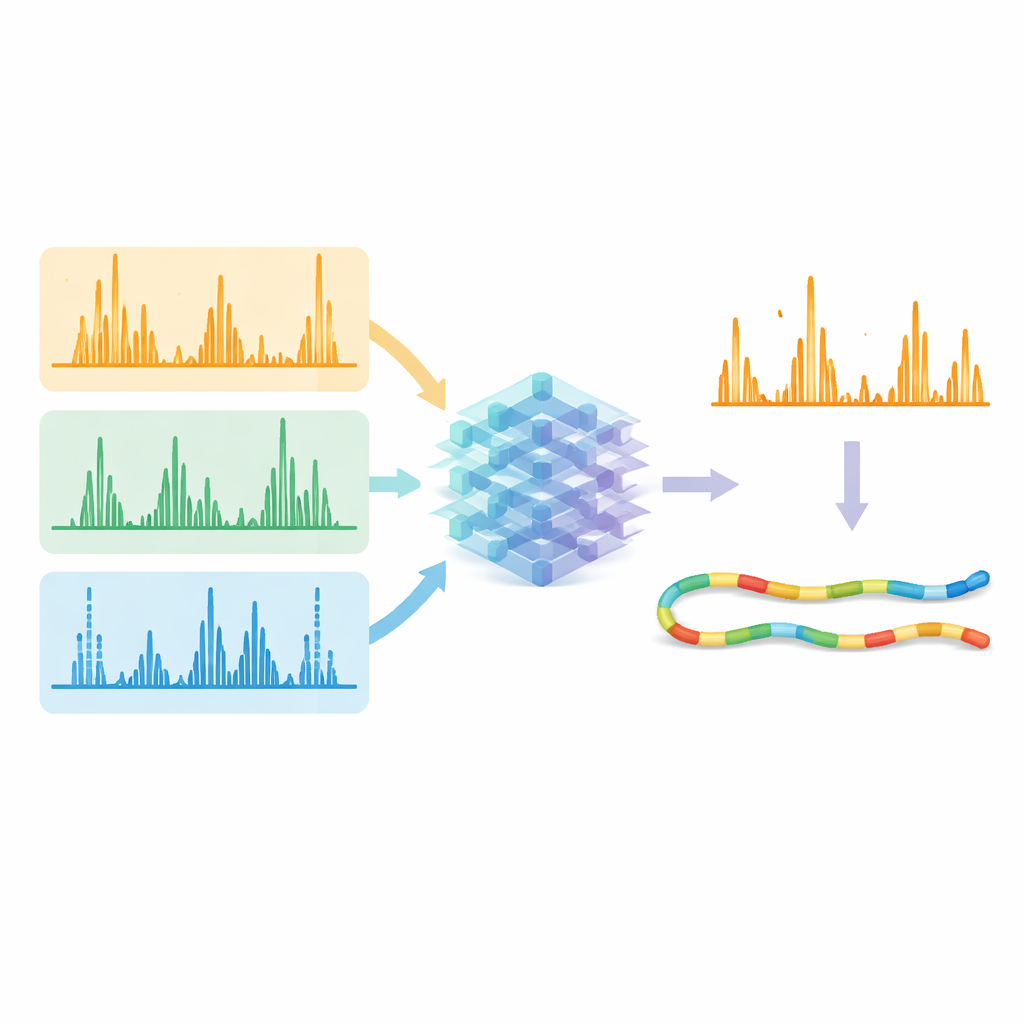
L’IA pulisce il segnale
La vera prova era se queste predizioni potessero migliorare il numero di peptidi identificati con sicurezza a partire dai dati grezzi. I ricercatori hanno fornito sia gli spettri misurati sia i modelli predetti dall’IA agli strumenti di ricerca e di ricalcolo esistenti. Quando hanno chiesto al software di concentrarsi sui frammenti che il modello indicava come forti e presenti, le corrispondenze corrette sono emerse in modo più netto rispetto a quelle false. Nei dati raccolti con diversi metodi di frammentazione ed enzimi, il numero di corrispondenze peptide–spettro identificate con sicurezza è aumentato tipicamente di oltre il 10% e, in alcuni casi difficili, di oltre il 30%. Importante, i metodi alternativi che usano elettroni e luce ultravioletta hanno ora raggiunto un’efficienza di identificazione simile al metodo standard basato sulle collisioni, offrendo al contempo una copertura di sequenza più ampia e informazioni più uniche sulle proteine.
Portare metodi avanzati nell’uso quotidiano
Poiché il modello di IA è liberamente disponibile ed è integrato nel software di spettrometria di massa più diffuso, può essere usato non solo per misure tradizionali e mirate ma anche per le più recenti strategie di acquisizione indipendente dai dati che scandagliano ampie porzioni del campione in una volta. Test su miscele di cellule umane, vegetali e batteriche hanno mostrato che il modello si generalizza bene tra le specie. In termini pratici, questo lavoro rimuove un ostacolo chiave che aveva mantenuto metodi di frammentazione potenti ma poco sfruttati confinati agli specialisti. Unificando diversi modi di frammentare le proteine sotto un unico modello predittivo, lo studio fornisce un percorso verso analisi di routine ad alta copertura di paesaggi proteici complessi, rendendo più semplice per i ricercatori individuare varianti rare, mappare modifiche e, in ultima analisi, ottenere un quadro più completo di come le proteine si comportano in salute e malattia.
Citazione: Levin, N., Saylan, C.C., Lapin, J. et al. Integration of alternative fragmentation techniques into standard LC-MS workflows using a single deep learning model enhances proteome coverage. Nat Methods 23, 805–814 (2026). https://doi.org/10.1038/s41592-026-03042-9
Parole chiave: proteomica, spettrometria di massa, deep learning, frammentazione delle proteine, predizione spettrale