Clear Sky Science · zh
REDD1–NF-κB–miRNAs–eNOS/SIRT1轴介导肥胖诱导的内皮细胞衰老与高血压
为何肥胖使我们的血管更快衰老
众所周知,肥胖会增加高血压、心肌梗死和中风的风险,但过多脂肪究竟如何使血管变得老化和僵硬一直不甚清晰。本研究揭示了动脉内薄薄细胞层内的一段分子“对话”,展示了肥胖引发的代谢应激如何使这些细胞过早衰老、损害肾脏并驱动高血压。理解这一系列事件指向了新的、非常具体的药物靶点,可能用于保护肥胖人群的血管。
血管内皮细胞承受的隐形压力
血管最内层由内皮细胞构成,负责调控血流、血管舒张和炎症。在肥胖状态下,血液中的脂肪酸、胆固醇、葡萄糖和类激素信号水平升高。作者通过给小鼠喂高脂饮食来模拟这种状态,并用与肥胖相关的因子(如棕榈酸、氧化低密度脂蛋白、高糖、瘦素和抗性素)处理人类内皮细胞。这些条件使细胞出现典型的衰老特征:永久性停止分裂、废物积累并释放促炎物质。在小鼠体内,主动脉——离开心脏的主动脉——出现了内皮细胞衰老斑块并丧失正常舒张能力,促成了血压升高。
将细胞推入衰老的应激传感器
研究组聚焦于一种响应应激的蛋白REDD1,它会被代谢性和氧化应激诱导。他们发现代谢性危险因素显著提升了培养的人和小鼠内皮细胞以及肥胖小鼠动脉中的REDD1水平。当在血管中实验性地增加REDD1时,内皮细胞迅速进入衰老状态,一氧化氮产生下降,动脉僵硬,从而驱动高血压。相反,一种无法通过关键炎症通路传递信号的REDD1突变体未能引起这些问题。基因缺失REDD1的小鼠,或仅在内皮细胞中缺失REDD1的小鼠在很大程度上受到保护:它们的动脉衰老细胞更少,保护性分子水平更高,血管舒张功能更好,尽管肥胖但血压较低。
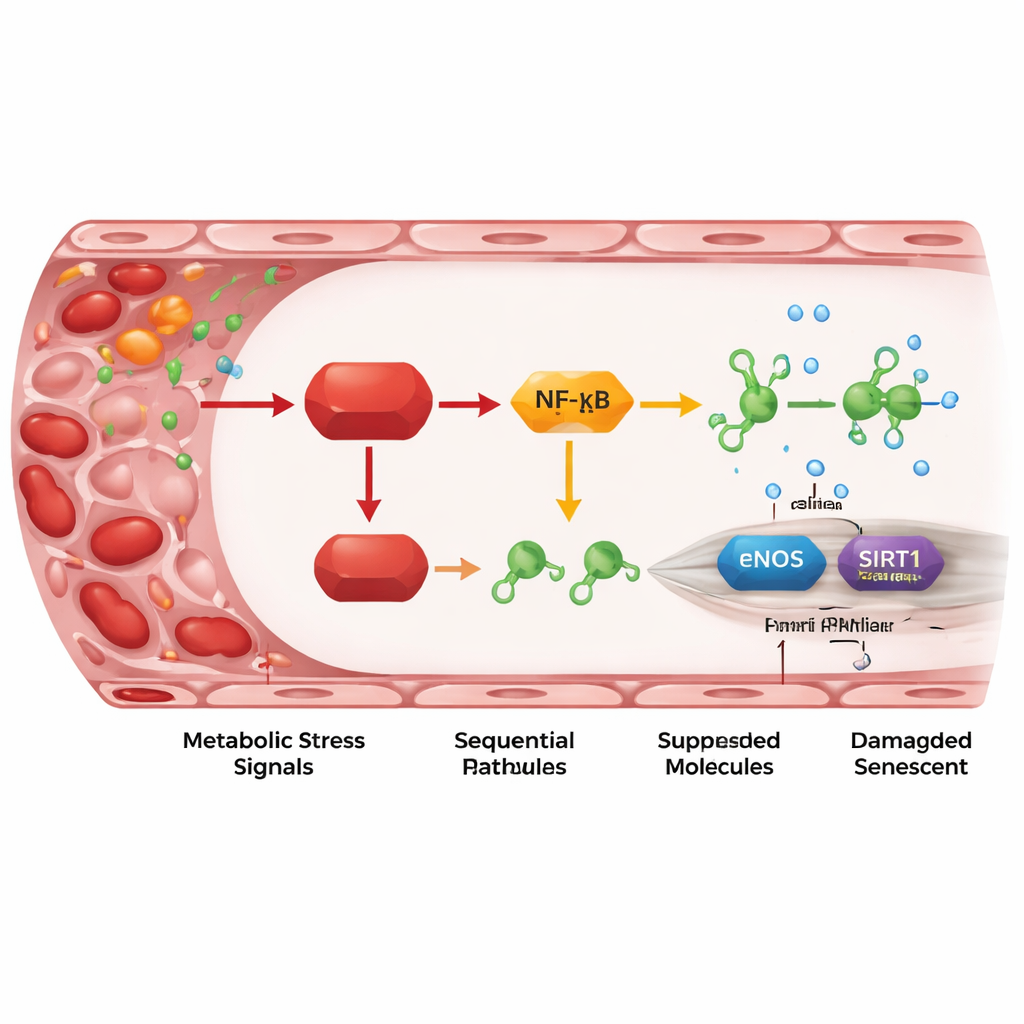
从REDD1到保护基因的炎性中继
深入研究后,研究者描绘出一条将REDD1与血管损伤相连的分子中继。REDD1激活了一种“非典型”的NF-κB通路——炎症的核心调控器。这进一步提高了两种小调控RNA的水平:miR-155-5p,尤其是miR-214-3p。这些微小RNA在内皮细胞内充当对两种保护性蛋白的微小制动器:eNOS产生一氧化氮以松弛血管,SIRT1则有助于维持细胞青春和DNA稳定性。当REDD1水平升高时,微RNA水平上升,eNOS和SIRT1下降,一氧化氮生成减少,衰老标志增加。阻断这些微RNA或提升SIRT1或一氧化氮可逆转大部分损伤,表明该轴不仅与血管壁的衰老相关联,而且确实驱动了这一过程。
来自基因模型与肾脏影响的证据
若干小鼠模型使作者能够在活体中验证该轴的每一步。缺失REDD1的肥胖小鼠、表达不可传导信号的REDD1突变体的小鼠或仅在内皮细胞中删除REDD1的小鼠都表现出减少的动脉衰老和较低的血压。缺失miR-214-3p的小鼠也获得类似保护:尽管肥胖时REDD1仍升高,eNOS和SIRT1水平仍较高,一氧化氮信号保存得更好,血管衰老和高血压被削弱。相同的REDD1–微RNA通路也影响肾脏,而肾脏对长期血压控制至关重要。在肥胖的野生型小鼠中,肾脏肿大、出现纤维化并有更多肌酐泄漏入血。这些肾损伤迹象在缺失REDD1、缺失内皮REDD1或缺失miR-214-3p的小鼠中要轻得多,提示保护血管也有助于保障肾功能。
这对未来疗法意味着什么
对外行来说,这项工作表明肥胖并非仅通过增加体重来推高血压;它在更深层次上重编程了血管内皮的生物学。研究识别出一条关键的罪魁链——REDD1、非典型NF-κB信号通路以及微RNA miR-155-5p和miR-214-3p——它们共同关闭保护性的eNOS和SIRT1,导致血管和肾脏更快衰老并升高血压。因为该链条的每一环在小鼠中都可以被干预以在肥胖存在后恢复较健康的血管,REDD1–NF-κB–微RNA–eNOS/SIRT1轴成为预防或治疗肥胖相关高血压及其并发症的有前景的药物蓝图。
引用: Choi, Y.K., Lee, DK., Park, M. et al. The REDD1–NF-κB–miRNAs–eNOS/SIRT1 axis mediates obesity-induced endothelial cell senescence and hypertension. Nat Commun 17, 3843 (2026). https://doi.org/10.1038/s41467-026-70601-1
关键词: 肥胖, 内皮细胞衰老, 高血压, 微小RNA, 一氧化氮