Clear Sky Science · en
The REDD1–NF-κB–miRNAs–eNOS/SIRT1 axis mediates obesity-induced endothelial cell senescence and hypertension
Why our blood vessels age faster in obesity
Obesity is well known for raising the risk of high blood pressure, heart attacks, and strokes, but exactly how excess fat makes our blood vessels old and stiff has remained murky. This study uncovers a molecular "conversation" inside the thin cell layer that lines our arteries, showing how metabolic stress from obesity can prematurely age these cells, damage the kidneys, and drive hypertension. Understanding this chain of events points to new, very specific targets for drugs that might protect blood vessels in people living with obesity.
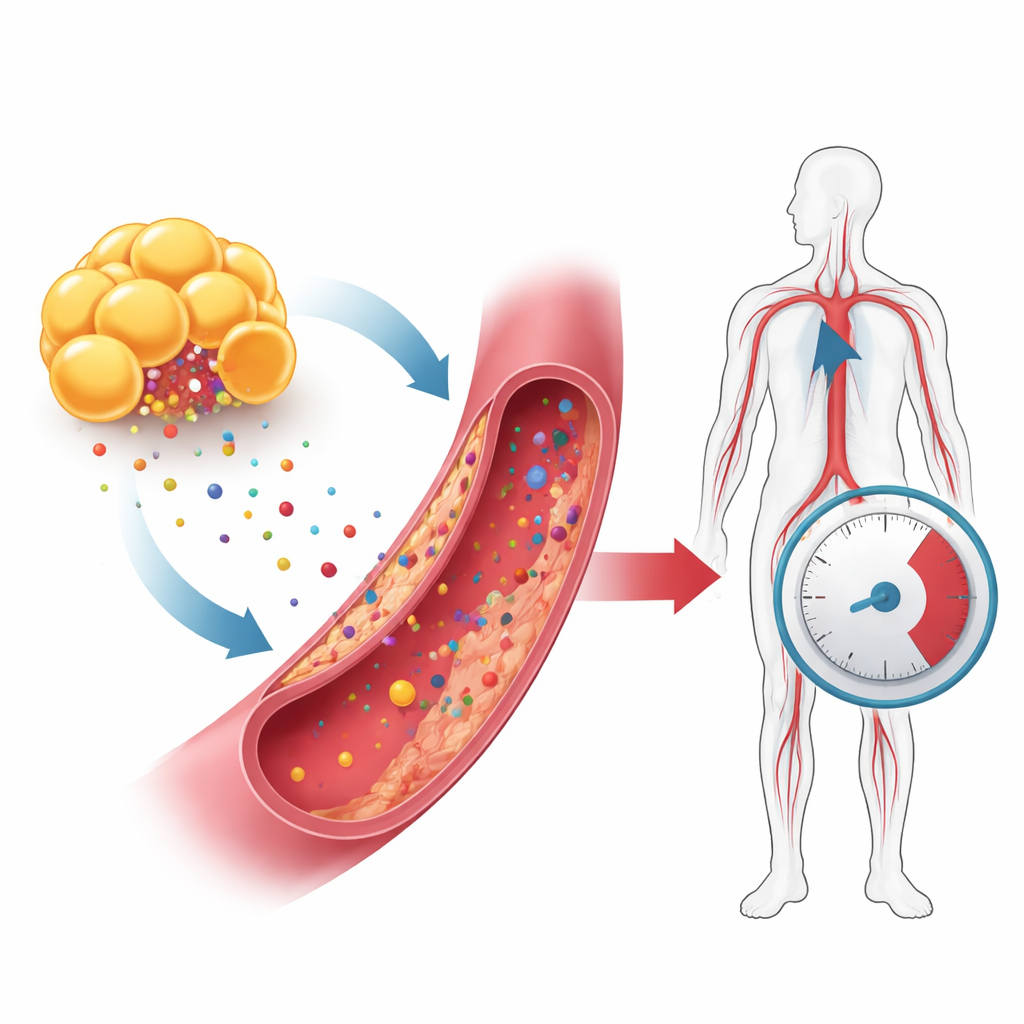
The hidden strain on vessel lining cells
The innermost lining of blood vessels is made of endothelial cells, which act as gatekeepers for blood flow, vessel relaxation, and inflammation. In obesity, levels of fatty acids, cholesterol, sugar, and hormone-like signals rise in the bloodstream. The authors fed mice a high-fat diet to mimic this state and treated human endothelial cells with obesity-related factors such as palmitic acid, oxidized LDL, high glucose, leptin, and resistin. These conditions caused the cells to show classic signs of senescence, a state of permanent arrest in which cells stop dividing, accumulate waste, and release inflammatory substances. In the mice, the aorta—the main artery leaving the heart—developed patches of aged endothelial cells and lost its ability to relax properly, contributing to increased blood pressure.
A stress sensor that tips cells into old age
The team focused on a stress-responsive protein called REDD1, which is switched on by metabolic and oxidative stress. They found that metabolic risk factors strongly boosted REDD1 levels in cultured human and mouse endothelial cells and in the arteries of obese mice. When REDD1 was experimentally increased in blood vessels, endothelial cells rapidly became senescent, nitric oxide production dropped, and arteries stiffened, driving hypertension. In contrast, a mutant form of REDD1 that could no longer signal through a key inflammatory pathway failed to cause these problems. Mice genetically lacking REDD1, or lacking REDD1 specifically in their endothelial cells, were largely protected: their arteries showed fewer senescent cells, higher levels of protective molecules, better vessel relaxation, and lower blood pressure despite obesity.
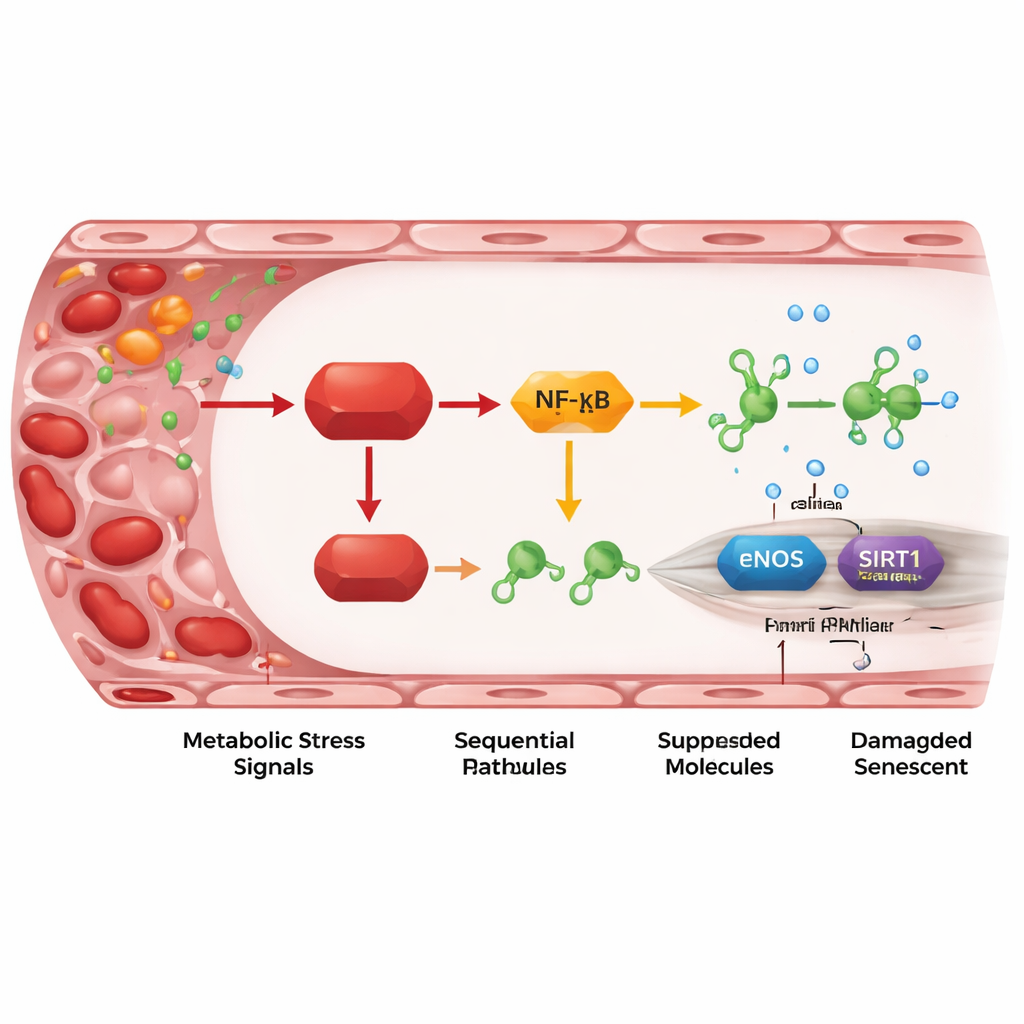
An inflammatory relay from REDD1 to protective genes
Digging deeper, the researchers mapped out a relay of molecular steps linking REDD1 to vascular damage. REDD1 activates an "atypical" version of the NF-κB pathway, a master controller of inflammation. This, in turn, raises levels of two small regulatory RNAs, miR-155-5p and especially miR-214-3p. These microRNAs act as tiny brakes on two protective proteins in endothelial cells: eNOS, which produces nitric oxide to relax vessels, and SIRT1, which helps maintain cellular youthfulness and DNA stability. When REDD1 was high, microRNA levels rose, eNOS and SIRT1 levels fell, nitric oxide production declined, and senescence markers increased. Blocking the microRNAs or boosting SIRT1 or nitric oxide could reverse much of the damage, showing that this axis is not just associated with, but actually drives, the aging process in the vessel wall.
Proof from genetic models and kidney effects
Several mouse models allowed the authors to test each step of this axis in living animals. Obese mice lacking REDD1, expressing the non-signaling REDD1 mutant, or engineered to delete REDD1 only in endothelial cells all showed reduced arterial aging and lower blood pressure. Mice that lacked miR-214-3p were similarly protected: even though REDD1 still rose with obesity, eNOS and SIRT1 remained higher, nitric oxide signaling was better preserved, and vascular senescence and hypertension were blunted. The same REDD1–microRNA pathway affected the kidneys, organs that are crucial for long-term blood pressure control. In obese wild-type mice, kidneys enlarged, became fibrotic, and leaked more creatinine into the blood. These signs of renal damage were much milder in mice lacking REDD1, lacking endothelial REDD1, or lacking miR-214-3p, suggesting that protecting blood vessels also helped safeguard kidney function.
What this means for future therapies
To a lay observer, this work shows that obesity does not simply push up blood pressure through extra body mass; it reprograms the biology of the vessel lining at a deep level. The study identifies a key chain of culprits—REDD1, atypical NF-κB signaling, and microRNAs miR-155-5p and miR-214-3p—that together shut down protective eNOS and SIRT1, causing blood vessels and kidneys to age faster and raising blood pressure. Because each link in this chain can be altered in mice to restore healthier vessels even after obesity is present, the REDD1–NF-κB–microRNA–eNOS/SIRT1 axis emerges as a promising blueprint for drugs aimed at preventing or treating obesity-linked hypertension and its complications.
Citation: Choi, Y.K., Lee, DK., Park, M. et al. The REDD1–NF-κB–miRNAs–eNOS/SIRT1 axis mediates obesity-induced endothelial cell senescence and hypertension. Nat Commun 17, 3843 (2026). https://doi.org/10.1038/s41467-026-70601-1
Keywords: obesity, endothelial senescence, hypertension, microRNA, nitric oxide