Clear Sky Science · tr
Fruktoz-2,6-bisfosfat, TDP-43 patolojisinin tetiklediği genoma onarım eksikliğini motor nöron hastalıklarında onarıyor
Sinir Hücrelerinde Kırık DNA Neden Önemli?
Amyotrofik lateral skleroz (ALS) ve frontotemporal demans (FTD), insanlardan hareket, konuşma ve hafızayı çalan yıkıcı beyin hastalıklarıdır. Yıllardır bilim insanları, TDP-43 adlı bir proteinin bu koşullarda sinir hücreleri içinde yanlış yerde birikme eğiliminde olduğunu biliyordu; ancak bunun nöron ölümüne nasıl yol açtığı belirsiz kaldı. Bu çalışma, enerji metabolizması ile hücrenin kırık DNA’yı onarma yeteneği arasında şaşırtıcı bir bağlantı ortaya koyuyor ve küçük bir şekerle ilişkili molekülün hem DNA hasarını hem de hastalık modellerindeki hareket bozukluklarını kısmen geri çevirebildiğini gösteriyor.
Hücresel Onarım Atölyesinde Trafik Sıkışması
Sinir hücreleri, özellikle hücrenin çalışması için aktif olarak okunan genlerde, sürekli DNA kırıklarıyla karşılaşır. Normalde PNKP adlı bir enzim, kırık DNA uçlarını temizleyerek onarımın tamamlanmasını sağlar. Yazarlar, ALS ve FTD’li kişilerin beyin dokusunda PNKP aktivitesinin keskin biçimde azaldığını; buna rağmen enzimin miktarının hâlâ mevcut olduğunu buldular. Aynı zamanda, aktif genlerde kalıcı DNA hasarına dair belirgin kanıtlar vardı; bu da onarım makinelerinin tam olarak nöron işlevini korumak için en çok ihtiyaç duyulan yerde tıkandığını gösteriyor.
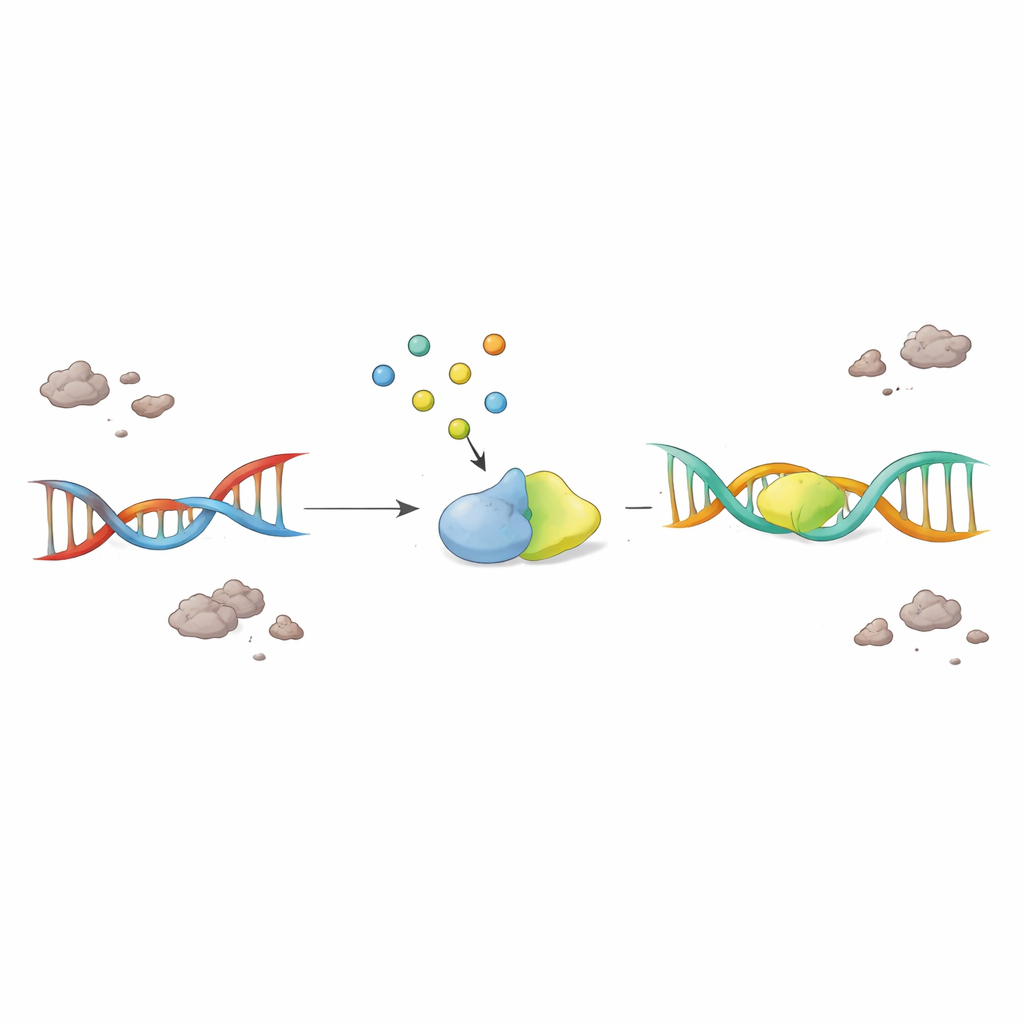
Yanlış Yere Yerleşen Bir Proteinin DNA Onarımını Nasıl Bozduğu
TDP-43 normalde DNA’nın saklandığı çekirdekte bulunur ve tehlikeli çift iplik kırıklarının onarımını koordine etmeye yardımcı olur. Ancak ALS ve FTD’de TDP-43 hücrenin dış bölgesine sızar ve yapışkan agregatlar oluşturur. Araştırmacılar kültür hücrelerinde TDP-43 seviyesini düşürdüklerinde, çekirdekteki TDP-43 kaybının PNKP’nin kırık DNA’yı işlemeye yeteneğini, PNKP miktarı değişmese bile bozmak için yeterli olduğunu gösterdiler. Ayrıca, normalde TDP-43, PNKP ve aktif genlerdeki DNA kırıklarını kapatan diğer faktörleri içeren daha geniş bir onarım ekibinin, TDP-43 yanlış konumlandığında parçalandığını gözlemlediler. Sonuç, özellikle yoğun kullanılan genlerde DNA hasarının birikmesi; bu da zaten savunmasız olan motor nöronları daha da hassas hale getiriyor.
Şeker Kullanımı ile Genom Koruması Arasındaki Eksik Bağlantı
Çalışma daha sonra bu onarım başarısızlığını metabolik bir anahtara bağladı. PNKP, verimli çalışmak için PFKFB3 enzimi tarafından üretilen fruktoz-2,6-bisfosfat (F2,6BP) adlı küçük bir moleküle bağımlıdır. ALS ve FTD beyin örneklerinde PFKFB3 proteini ve F2,6BP düzeyleri belirgin şekilde azalmışken, PFKFB3 için genetik talimatlar büyük ölçüde sağlam kaldı. Bu durum, üretimle ilgili bir sorundan ziyade PFKFB3 proteininin artmış yıkımına işaret ediyor. Hastadan türetilmiş sinir öncül hücrelerde ve TDP-43 patolojisini taklit eden birkaç farklı hayvan modelinde aynı desen ortaya çıktı: yanlış yerleşmiş TDP-43, düşük PFKFB3, zayıf PNKP aktivitesi ve aktif genlerde artmış DNA hasarı.
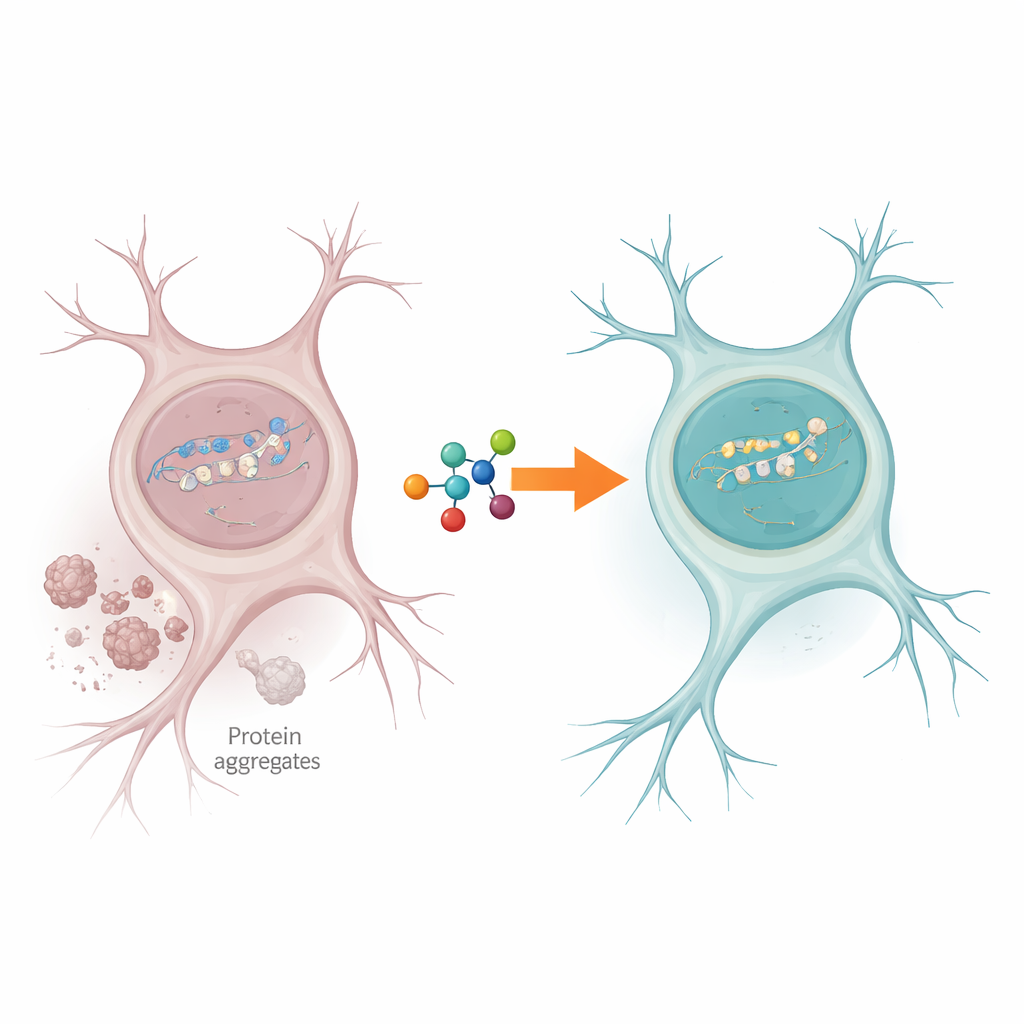
Hastalıklı Nöronları Kurtarmak İçin Küçük Bir Molekülün Güçlendirilmesi
Önemli olarak, bilim insanları F2,6BP’yi geri eklemenin tıkanmış onarım sistemini yeniden canlandırıp canlandırmayacağını test ettiler. ALS ve FTD beyinlerinden alınan nükleer ekstreye F2,6BP verildiğinde, PNKP aktivitesi doz bağımlı olarak geri döndü; buna karşın yakından ilişkili şeker molekülleri hiçbir etki göstermedi. Hastadan türetilmiş ve hastalıkla ilişkili TDP-43 mutasyonları taşıyan hücrelerde F2,6BP hem PNKP aktivitesini geri kazandırdı hem de sitozoldeki anormal, agregatlaşmış TDP-43 miktarını azalttı. TDP-43 patolojili bir fare modelinde F2,6BP yine PNKP fonksiyonunu canlandırdı. En çarpıcı biçimde, motor nöronlarda mutant insan TDP-43 ifade eden bir meyve sineği modelinde ağız yoluyla verilen F2,6BP, sineklerin tırmanma yeteneğini iyileştirdi ve kırılmaya duyarlı, etkinlik yoğunluğu yüksek büyük genlerdeki DNA hasarını azalttı.
Gelecekteki Tedaviler İçin Ne Anlama Gelebilir?
Bu bulgular, TDP-43 patolojisinin enerji metabolizmasını DNA onarımıyla bağlayan kritik bir ekseni aksattığı bir tablo çiziyor: PFKFB3 F2,6BP üretir, F2,6BP PNKP’yi besler ve PNKP aktif genleri kırık birikimine karşı korur. Bu eksen başarısız olduğunda, nöronlar giderek genomik kararlılıklarını ve işlevlerini kaybeder. F2,6BP’yi yeniden sağlayarak yazarlar PNKP aktivitesini canlandırabildiler, DNA onarımını daha verimli hale getirdiler ve canlı bir organizmadaki hareket sorunlarını hafifletti. Bunları insan tedavilerine dönüştürmek ve metabolik etkileri hassaslaştırmak için daha yapılması gereken çok iş olsa da çalışma, bu metabolik–onarım yolunu dikkatli şekilde hedeflemenin ALS, FTD ve muhtemelen TDP-43 protein birikimiyle işaretli diğer bozukluklar için yeni, mekanizma temelli bir tedavi yolu açabileceğini öne sürüyor.
Atıf: Chakraborty, A., Mitra, J., Malojirao, V.H. et al. Fructose-2,6-bisphosphate restores TDP-43 pathology-driven genome repair deficiency in motor neuron diseases. Commun Biol 9, 563 (2026). https://doi.org/10.1038/s42003-026-09787-5
Anahtar kelimeler: ALS, TDP-43, DNA onarımı, nörodejenerasyon, hücre metabolizması