Clear Sky Science · en
Fructose-2,6-bisphosphate restores TDP-43 pathology-driven genome repair deficiency in motor neuron diseases
Why Broken DNA Matters in Nerve Cells
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are devastating brain diseases that rob people of movement, speech, and memory. For years, scientists have known that a protein called TDP-43 clumps in the wrong place inside nerve cells in these conditions, but how this leads to neuron death has remained murky. This study uncovers a surprising link between energy metabolism and the cell’s ability to repair broken DNA, and shows that a small sugar-related molecule can partly reverse both DNA damage and movement problems in disease models.
A Traffic Jam in the Cell’s Repair Shop
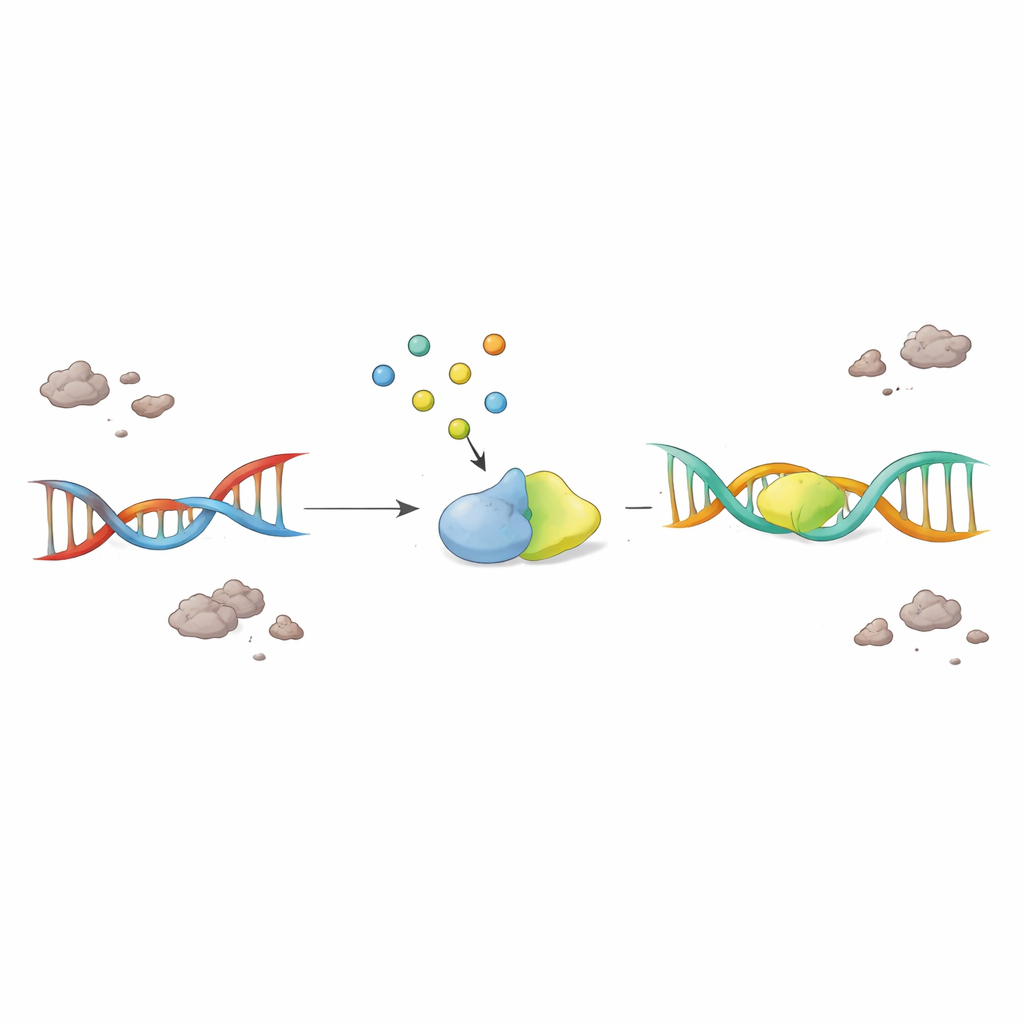
Nerve cells constantly experience DNA breaks, especially in genes that are actively read to keep the cell running. Normally, an enzyme called PNKP helps tidy up the loose ends of broken DNA so that repair can be completed. The authors found that in brain tissue from people with ALS and FTD, PNKP’s activity was sharply reduced, even though the enzyme itself was still present. At the same time, there was clear evidence of lingering DNA damage in active genes, suggesting that the repair machinery was stalled right where it is most needed to preserve neuron function.
How a Misplaced Protein Disrupts DNA Repair
TDP-43 usually resides in the nucleus, where DNA is stored, and helps coordinate the repair of dangerous double-strand breaks. In ALS and FTD, however, TDP-43 leaks into the cell’s outer region and forms sticky aggregates. By lowering TDP-43 levels in cultured cells, the researchers showed that this loss of nuclear TDP-43 is enough to cripple PNKP’s ability to process broken DNA, even though PNKP’s amount does not change. They also observed that a larger repair team, which normally includes TDP-43, PNKP, and other factors that seal DNA breaks in active genes, falls apart when TDP-43 is mislocalized. The result is a build-up of DNA damage specifically in heavily used genes, making already vulnerable motor neurons even more fragile.
A Missing Link Between Sugar Use and Genome Protection
The study then traces this repair failure back to a metabolic switch. PNKP depends on a small molecule called fructose-2,6-bisphosphate (F2,6BP), produced by the enzyme PFKFB3, to function efficiently. In ALS and FTD brain samples, PFKFB3 protein and F2,6BP levels were markedly reduced, while the genetic instructions for PFKFB3 were largely intact. This points to increased breakdown of the PFKFB3 protein rather than a problem with its production. In patient-derived nerve precursor cells and several different animal models that mimic TDP-43 pathology, the same pattern emerged: mislocalized TDP-43, lower PFKFB3, weaker PNKP activity, and more DNA damage in active genes.
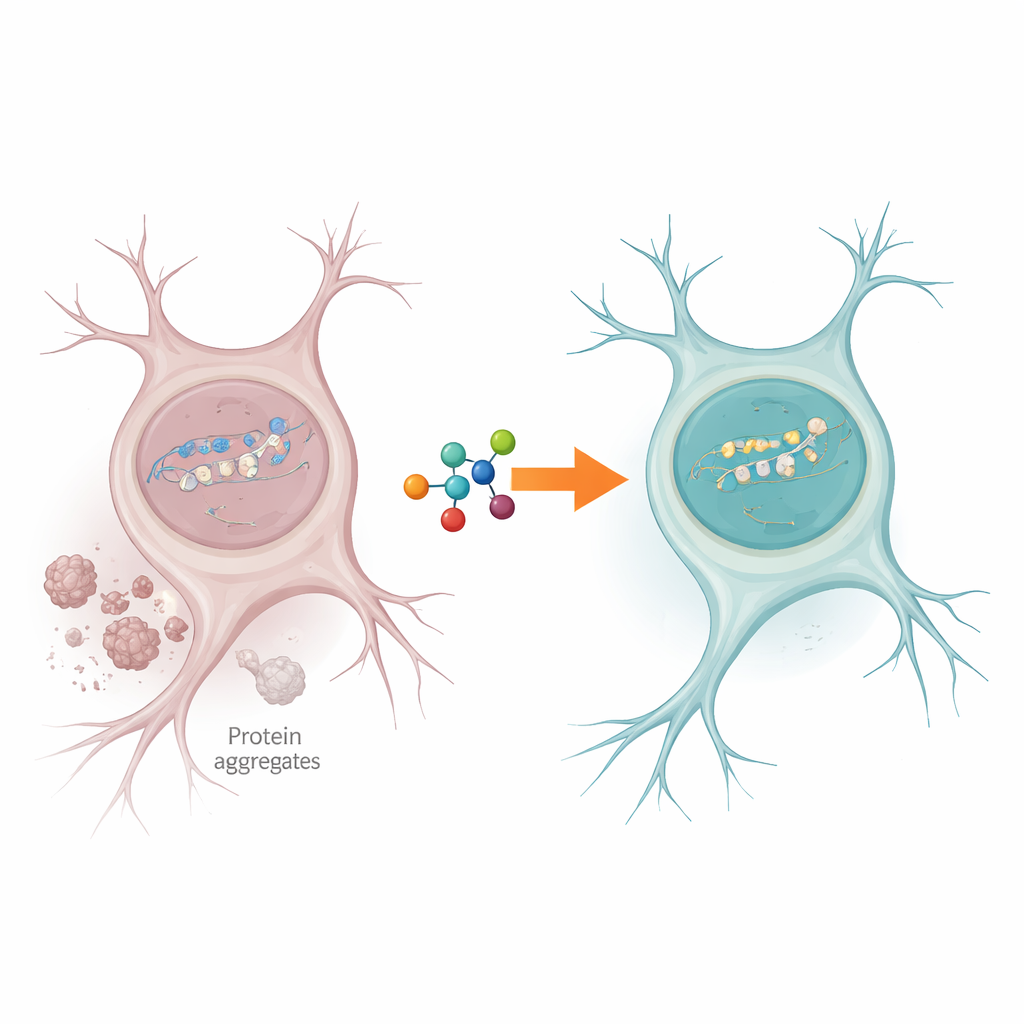
Boosting a Small Molecule to Rescue Sick Neurons
Crucially, the scientists tested whether simply adding back F2,6BP could reawaken the stalled repair system. When F2,6BP was supplied to nuclear extracts from ALS and FTD brains, PNKP activity rebounded in a dose-dependent manner, while closely related sugar molecules had no effect. In patient-derived cells carrying disease-linked TDP-43 mutations, F2,6BP both restored PNKP activity and reduced the amount of abnormal, aggregated TDP-43 in the cytosol. In a mouse model with TDP-43 pathology, F2,6BP again revived PNKP function. Most strikingly, in a fruit fly model expressing mutant human TDP-43 in motor neurons, oral F2,6BP treatment improved the flies’ climbing ability and reduced DNA damage in large, activity-heavy genes that are sensitive to breakage.
What This Could Mean for Future Treatments
Together, these findings paint a picture in which TDP-43 pathology derails a critical axis linking energy metabolism to DNA repair: PFKFB3 produces F2,6BP, F2,6BP fuels PNKP, and PNKP protects active genes from accumulating breaks. When this axis fails, neurons gradually lose genomic stability and function. By replenishing F2,6BP, the authors were able to revive PNKP activity, repair DNA more efficiently, and ease movement problems in a living organism. While much work remains to translate this into human therapies and to fine-tune metabolic effects, the study suggests that carefully targeting this metabolic–repair pathway could open a new, mechanism-based route to treating ALS, FTD, and possibly other disorders marked by TDP-43 protein buildup.
Citation: Chakraborty, A., Mitra, J., Malojirao, V.H. et al. Fructose-2,6-bisphosphate restores TDP-43 pathology-driven genome repair deficiency in motor neuron diseases. Commun Biol 9, 563 (2026). https://doi.org/10.1038/s42003-026-09787-5
Keywords: ALS, TDP-43, DNA repair, neurodegeneration, cell metabolism