Clear Sky Science · he
פרוקטוזה-2,6-ביספוספט משחזרת את חוסר היכולת לתקן גנום המונע על ידי פתולוגיית TDP-43 במחלות תאי עצב מוטוריים
מדוע DNA שבור חשוב בתאי עצב
נוירונל מוטורית צדית שיתוקית (ALS) ודמנציה פרונטוטמפורלית (FTD) הן מחלות מוח הרסניות שגוזלות מאנשים את היכולת לנוע, לדבר ולזכור. במשך שנים מדענים ידעו שחלבון בשם TDP-43 מהווה אגירות במקום הלא נכון בתוך תאי העצב במצבים אלה, אך איך זה מוביל למות הנוירונים נותר מעורפל. המחקר חושף קישור מפתיע בין מטבוליזם האנרגיה ליכולת התא לתקן DNA שבור, ומראה שמולקולה קטנה הקשורה לסוכר יכולה להחזיר חלקית גם את נזקי ה‑DNA וגם את הבעיות בתנועה במודלים של המחלה.
פקקי תנועה במפעל התיקון של התא
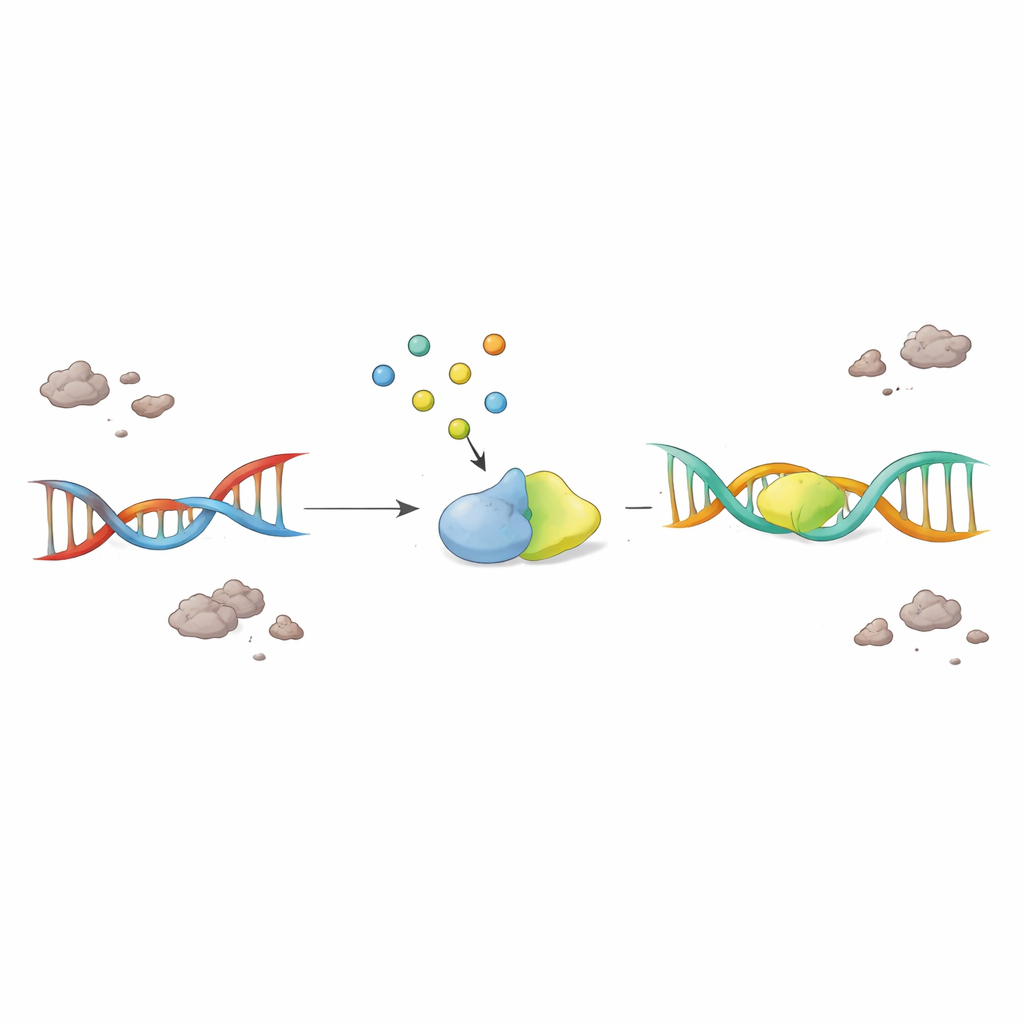
תאי עצב חווים באופן שוטף שברים ב‑DNA, במיוחד בגנים שנקראים באופן פעיל כדי לשמור על פעילות התא. בדרך כלל אנזים בשם PNKP עוזר לסדר את הקצוות הרופפים של ה‑DNA השבור כך שהתיקון יוכל להסתיים. החוקרים מצאו ברקמות מוח של חולי ALS ו‑FTD שפעילות ה‑PNKP ירדה באופן חד, אף על פי שהאנזים עצמו עדיין נוכח. באותו הזמן נצפו עדויות לנזקי DNA שטרם תוקנו בגנים פעילים, מה שמרמז שמכונת התיקון התקעה במקום שבו היא נדרשת ביותר לשמירה על תפקוד הנוירון.
איך חלבון ממוקם במקום הלא נכון מחריב את תיקון ה‑DNA
TDP-43 בדרך כלל מיושב בגרעין, שם מאוחסן ה‑DNA, ועוזר לתאם את תיקון שברים דו‑גדיליים מסוכנים. ב‑ALS ו‑FTD, לעומת זאת, TDP-43 דולף לציטופלסמה ויוצר אגירות דביקות. בהורדת רמות TDP-43 בתאים מועברים לגידול בעבדה, הראו החוקרים שהיעדרות ה‑TDP-43 הגרעיני מספיקה לשתק את יכולתו של PNKP לעבד DNA שבור, אף על פי שכמות ה‑PNKP לא השתנתה. הם גם ראו שקבוצת תיקון רחבה יותר, שבדרך כלל כוללת את TDP-43, PNKP ופקטורים אחרים שאוטמים שברי DNA בגנים פעילים, מתפוררת כש‑TDP-43 ממוקם במקום הלא נכון. התוצאה היא הצטברות נזקי DNA במיוחד בגנים בשימוש כבד, מה שהופך נוירונים מוטוריים שכבר פגיעים לפגיעים עוד יותר.
קשר חסר בין שימוש בסוכר להגנה על הגנום
המחקר מזהה את כישלון התיקון הזה כבעיה מטבולית. PNKP תלוי במולקולה קטנה בשם פרוקטוזה‑2,6‑ביספוספט (F2,6BP), שמיוצרת על ידי האנזים PFKFB3, כדי לתפקד ביעילות. בדגימות מוח של ALS ו‑FTD, חלבון PFKFB3 ורמות F2,6BP היו מוקטן בצורה בולטת, בעוד שהוראות הגנטיות ל‑PFKFB3 נשארו ברובן שלמות. ממצא זה מצביע על פירוק מוגבר של חלבון PFKFB3 ולא על בעיה בייצורו. בתאים מוקדמי עצב שמקורם בחולים ובכמה דגמי בעלי חיים המדמים פתולוגיית TDP-43, הופיע אותו דפוס: TDP-43 ממוקם לא נכון, PFKFB3 נמוך יותר, פעילות PNKP חלשה יותר, ויותר נזקי DNA בגנים פעילים.
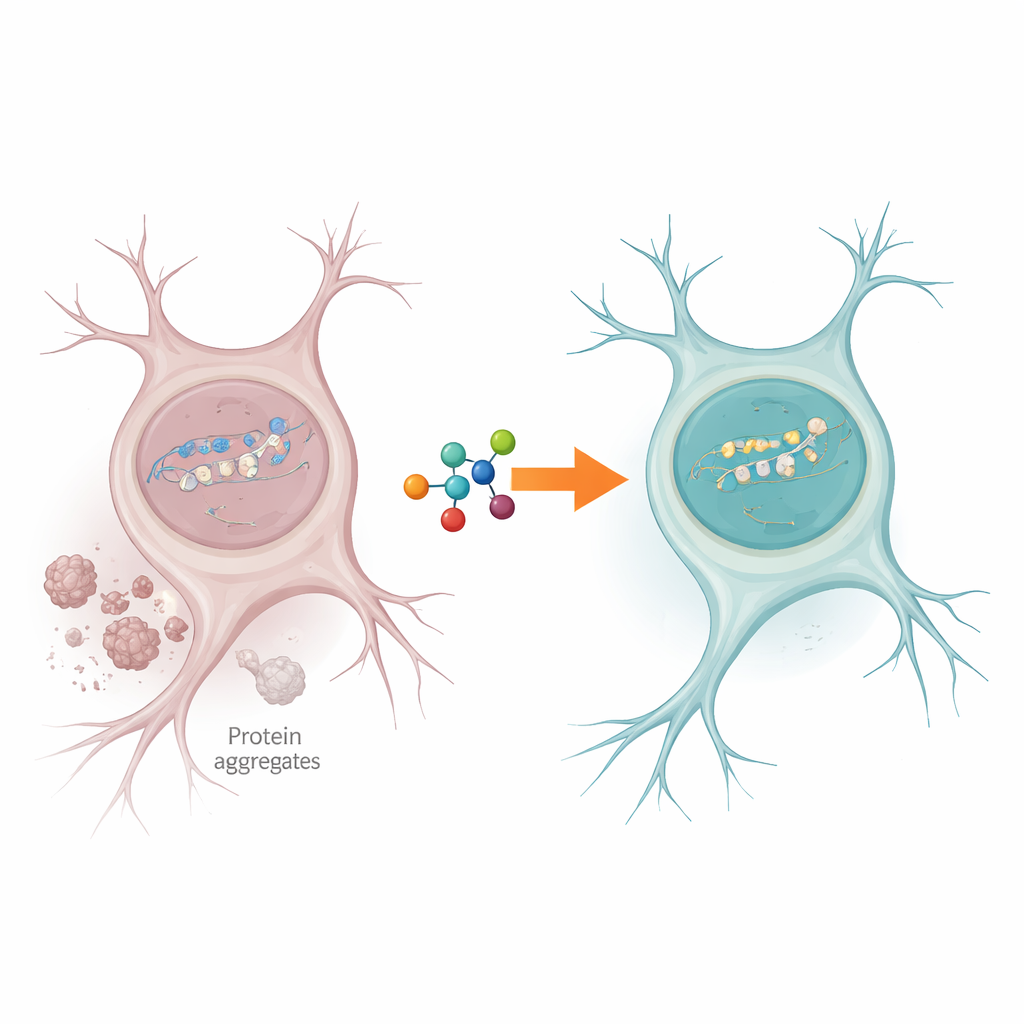
הגברה של מולקולה קטנה כדי להציל נוירונים חולים
חשוב מכך, המדענים בחנו האם הוספה פשוטה של F2,6BP יכולה להפעיל מחדש את מערכת התיקון התקועה. כאשר הוסיפו F2,6BP לתמציות גרעיניות מדגימות מוח של ALS ו‑FTD, פעילות PNKP חזרה במידה תלויה במינון, בעוד שמולקולות סוכר קרובות לא השפיעו. בתאים ממקור חולים הנושאים מוטציות ב‑TDP-43, F2,6BP שיקמה גם את פעילות PNKP והקטינה את כמות ה‑TDP-43 החריגה והאגורה בציטוזול. במודל עכבר עם פתולוגיית TDP-43, F2,6BP השיבה שוב את פעולת PNKP. ובהברות ביותר, במודל זבוב פירות שמביע TDP-43 אנושי מוטנט בנוירונים מוטוריים, טיפול אוראלי ב‑F2,6BP שיפר את יכולת הטיפוס של הזבובים והקטין את נזקי ה‑DNA בגנים גדולים ופעילים שרגישים לשבר.
מה זה עשוי לומר על טיפולים עתידיים
ביחד, הממצאים מציירים תמונה שבה פתולוגיית TDP-43 תוקעת ציר קריטי שמקשר בין מטבוליזם אנרגיה לתיקון DNA: PFKFB3 מייצר F2,6BP, F2,6BP מזין את PNKP, ו‑PNKP מגן על גנים פעילים מהצטברות שברים. כאשר ציר זה נכשל, הנוירונים מאבדים בהדרגה יציבות גנומית ותפקוד. על ידי השבתת מלאי ה‑F2,6BP, המחברים הצליחו להחזיר פעילות PNKP, לתקן DNA ביעילות רבה יותר ולהקל על בעיות תנועה באורגניזם חי. אמנם נדרש עוד עבודה רבה כדי לתרגם זאת לטיפולים בבני אדם ולכוון באופן מדויק את ההשפעות המטבוליות, המחקר מציע כי מיקוד זהיר בדרך המטבולית‑תיקונית יכול לפתוח מסלול חדש מבוסס מנגנון לטיפול ב‑ALS, FTD ואולי בהפרעות נוספות המאופיינות בהצטברות חלבון TDP-43.
ציטוט: Chakraborty, A., Mitra, J., Malojirao, V.H. et al. Fructose-2,6-bisphosphate restores TDP-43 pathology-driven genome repair deficiency in motor neuron diseases. Commun Biol 9, 563 (2026). https://doi.org/10.1038/s42003-026-09787-5
מילות מפתח: ALS, TDP-43, תיקון DNA, ניורודגנרציה, מטבוליזם תאאי