Clear Sky Science · es
La fructosa-2,6-bisfosfato restaura la deficiencia en la reparación del genoma provocada por la patología de TDP-43 en las enfermedades de motoneurona
Por qué importa el ADN roto en las células nerviosas
La esclerosis lateral amiotrófica (ELA) y la demencia frontotemporal (DFT) son enfermedades cerebrales devastadoras que privan a las personas del movimiento, el habla y la memoria. Durante años, los científicos han sabido que una proteína llamada TDP-43 se agrupa en el lugar equivocado dentro de las células nerviosas en estas afecciones, pero cómo esto conduce a la muerte neuronal ha seguido siendo oscuro. Este estudio descubre un vínculo sorprendente entre el metabolismo energético y la capacidad de la célula para reparar el ADN roto, y muestra que una pequeña molécula relacionada con los azúcares puede revertir en parte tanto el daño en el ADN como los problemas de movimiento en modelos de la enfermedad.
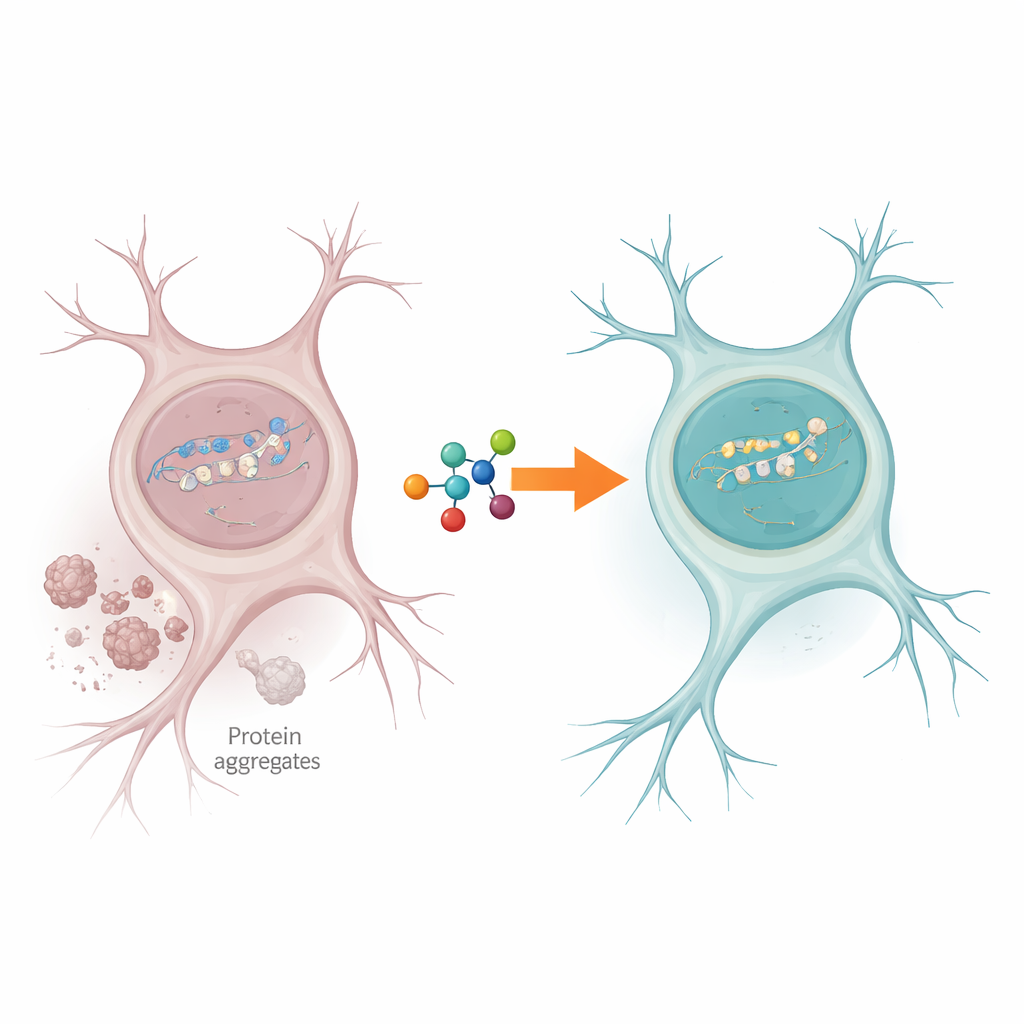
Un atasco en el taller de reparación de la célula
Las células nerviosas experimentan constantemente roturas del ADN, especialmente en genes que se leen activamente para mantener la célula en funcionamiento. Normalmente, una enzima llamada PNKP ayuda a ordenar los extremos sueltos del ADN roto para que la reparación pueda completarse. Los autores encontraron que en tejido cerebral de personas con ELA y DFT, la actividad de PNKP estaba marcadamente reducida, aunque la enzima en sí seguía presente. Al mismo tiempo, había evidencia clara de daño persistente en el ADN de genes activos, lo que sugiere que la maquinaria de reparación se atascaba justo donde más se necesita para preservar la función neuronal.
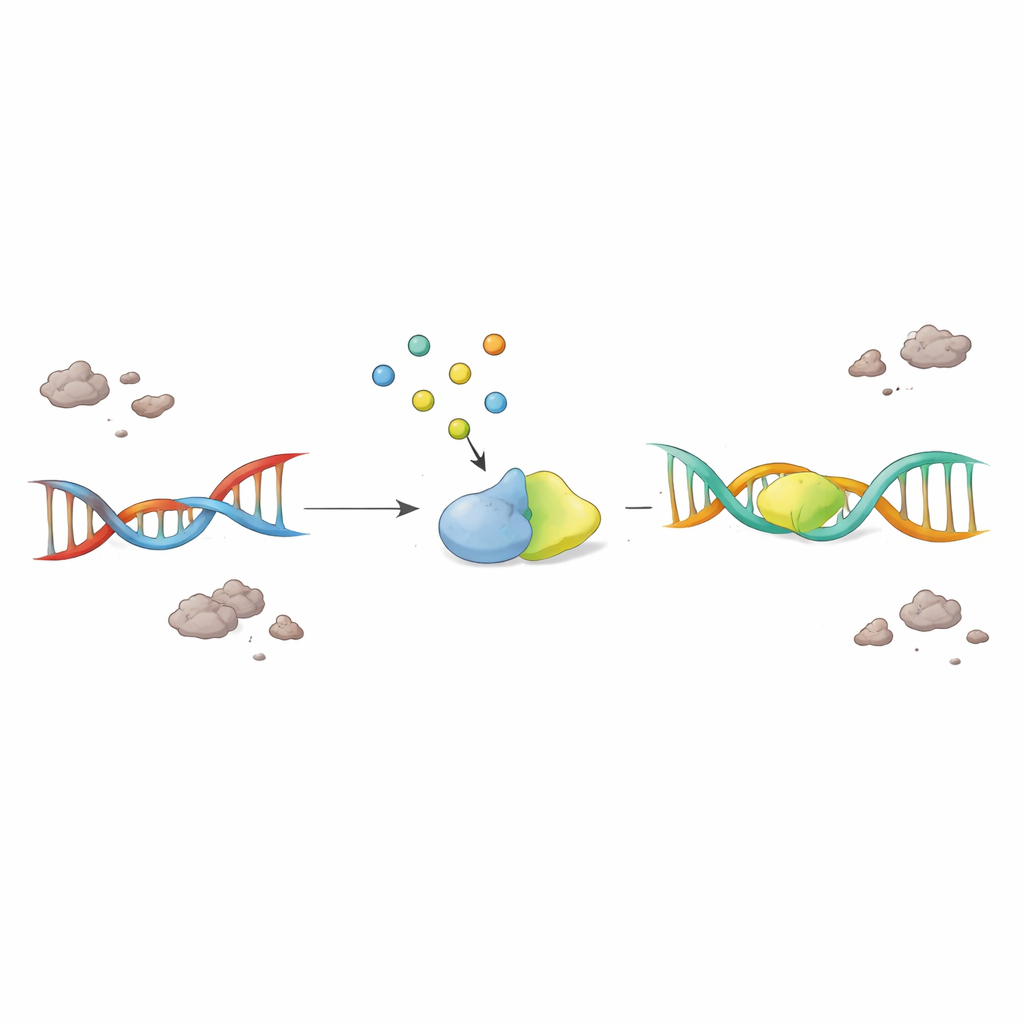
Cómo una proteína desplazada altera la reparación del ADN
TDP-43 normalmente reside en el núcleo, donde se almacena el ADN, y ayuda a coordinar la reparación de roturas de doble cadena peligrosas. En la ELA y la DFT, sin embargo, TDP-43 se filtra hacia la región externa de la célula y forma agregados pegajosos. Al reducir los niveles de TDP-43 en células en cultivo, los investigadores demostraron que esta pérdida de TDP-43 nuclear es suficiente para dejar a PNKP incapacitada para procesar el ADN roto, pese a que la cantidad de PNKP no cambia. También observaron que un equipo de reparación mayor, que normalmente incluye a TDP-43, PNKP y otros factores que sellan las roturas del ADN en genes activos, se desintegra cuando TDP-43 está mal localizado. El resultado es una acumulación de daño en el ADN específicamente en genes muy utilizados, lo que hace que las motoneuronas ya vulnerables sean aún más frágiles.
Un eslabón perdido entre el uso de azúcares y la protección del genoma
El estudio rastrea luego este fallo de reparación hasta un cambio metabólico. PNKP depende de una pequeña molécula llamada fructosa-2,6-bisfosfato (F2,6BP), producida por la enzima PFKFB3, para funcionar de forma eficiente. En muestras cerebrales de ELA y DFT, las proteínas PFKFB3 y los niveles de F2,6BP estaban notablemente reducidos, mientras que las instrucciones genéticas para PFKFB3 estaban en gran medida intactas. Esto apunta a un aumento de la degradación de la proteína PFKFB3 más que a un problema en su producción. En células precursoras nerviosas derivadas de pacientes y en varios modelos animales diferentes que imitan la patología de TDP-43, emergió el mismo patrón: TDP-43 mal localizado, menor PFKFB3, actividad más débil de PNKP y más daño en el ADN de genes activos.
Potenciar una pequeña molécula para rescatar neuronas enfermas
De forma crucial, los científicos probaron si simplemente devolver F2,6BP podía reactivar el sistema de reparación atascado. Cuando se suministró F2,6BP a extractos nucleares de cerebros con ELA y DFT, la actividad de PNKP repuntó de manera dependiente de la dosis, mientras que moléculas azucaradas estrechamente relacionadas no tuvieron efecto. En células derivadas de pacientes que portaban mutaciones de TDP-43 vinculadas a la enfermedad, F2,6BP restauró tanto la actividad de PNKP como redujo la cantidad de TDP-43 anómalo y agregado en el citosol. En un modelo de ratón con patología de TDP-43, F2,6BP volvió a activar la función de PNKP. Lo más llamativo fue que, en un modelo de mosca de la fruta que expresa TDP-43 humano mutante en las motoneuronas, el tratamiento oral con F2,6BP mejoró la capacidad de trepar de las moscas y redujo el daño en el ADN en genes grandes y muy activos que son sensibles a las roturas.
Qué podría significar esto para futuros tratamientos
En conjunto, estos hallazgos dibujan un panorama en el que la patología de TDP-43 descarrila un eje crítico que conecta el metabolismo energético con la reparación del ADN: PFKFB3 produce F2,6BP, F2,6BP alimenta a PNKP y PNKP protege a los genes activos de acumular roturas. Cuando este eje falla, las neuronas pierden gradualmente la estabilidad genómica y la función. Al reponer F2,6BP, los autores lograron reactivar la actividad de PNKP, reparar el ADN con mayor eficiencia y aliviar los problemas de movimiento en un organismo vivo. Aunque queda mucho trabajo por hacer para traducir esto en terapias humanas y afinar los efectos metabólicos, el estudio sugiere que dirigirse con precisión a esta vía metabólico-reparadora podría abrir una nueva vía basada en mecanismos para tratar la ELA, la DFT y posiblemente otros trastornos caracterizados por la acumulación de la proteína TDP-43.
Cita: Chakraborty, A., Mitra, J., Malojirao, V.H. et al. Fructose-2,6-bisphosphate restores TDP-43 pathology-driven genome repair deficiency in motor neuron diseases. Commun Biol 9, 563 (2026). https://doi.org/10.1038/s42003-026-09787-5
Palabras clave: ELA, TDP-43, reparación del ADN, neurodegeneración, metabolismo celular