Clear Sky Science · it
Fruttosio-2,6-bisfosfato ripristina la carenza di riparazione del genoma indotta dalla patologia di TDP-43 nelle malattie dei motoneuroni
Perché il DNA rotto è importante nelle cellule nervose
La sclerosi laterale amiotrofica (SLA) e la demenza frontotemporale (FTD) sono malattie cerebrali devastanti che privano le persone del movimento, della parola e della memoria. Per anni gli scienziati hanno osservato che una proteina chiamata TDP-43 si aggrega nel posto sbagliato all’interno delle cellule nervose in queste condizioni, ma il modo in cui ciò conduce alla morte neuronale è rimasto poco chiaro. Questo studio scopre un collegamento sorprendente tra il metabolismo energetico e la capacità della cellula di riparare il DNA rotto, e mostra che una piccola molecola legata agli zuccheri può in parte invertire sia il danno al DNA sia i problemi di movimento nei modelli di malattia.
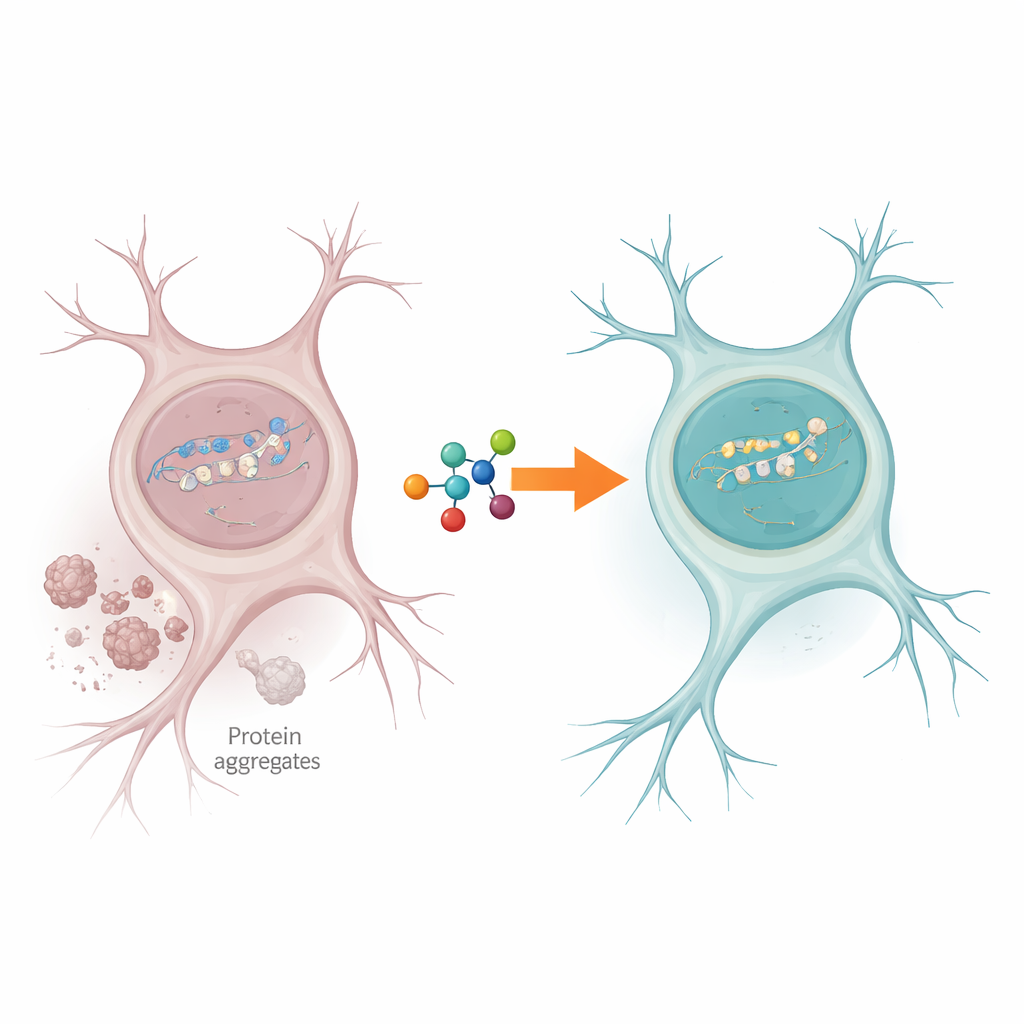
Un ingorgo nell’officina di riparazione della cellula
Le cellule nervose sperimentano costantemente rotture del DNA, specialmente nei geni che vengono attivamente letti per mantenere il funzionamento cellulare. Normalmente, un enzima chiamato PNKP aiuta a sistemare le estremità libere del DNA rotto in modo che la riparazione possa essere completata. Gli autori hanno rilevato che nei tessuti cerebrali di persone con SLA e FTD l’attività di PNKP era fortemente ridotta, anche se l’enzima stesso era ancora presente. Allo stesso tempo, c’erano chiari segni di danno al DNA persistente nei geni attivi, suggerendo che la macchina di riparazione era bloccata proprio dove è più necessaria per preservare la funzione neuronale.
Come una proteina spostata interrompe la riparazione del DNA
TDP-43 risiede di norma nel nucleo, dove è conservato il DNA, e aiuta a coordinare la riparazione delle pericolose rotture a doppio filamento. Nella SLA e nella FTD, tuttavia, TDP-43 fuoriesce nella regione citoplasmatica della cellula e forma aggregati appiccicosi. Riducendo i livelli di TDP-43 in cellule coltivate, i ricercatori hanno dimostrato che questa perdita di TDP-43 nucleare è sufficiente a compromettere la capacità di PNKP di processare il DNA rotto, anche se la quantità di PNKP non cambia. Hanno inoltre osservato che un più ampio complesso di riparazione, che normalmente include TDP-43, PNKP e altri fattori che sigillano le rotture del DNA nei geni attivi, si disgrega quando TDP-43 è mal localizzato. Il risultato è un accumulo di danni al DNA specificamente nei geni molto utilizzati, rendendo i motoneuroni già vulnerabili ancora più fragili.
Un anello mancante tra l’uso degli zuccheri e la protezione del genoma
Lo studio ricostruisce poi questo fallimento della riparazione fino a un interruttore metabolico. PNKP dipende da una piccola molecola chiamata fruttosio-2,6-bisfosfato (F2,6BP), prodotta dall’enzima PFKFB3, per funzionare in modo efficiente. Nei campioni cerebrali di SLA e FTD, le proteine PFKFB3 e i livelli di F2,6BP risultavano marcatamente ridotti, mentre le istruzioni genetiche per PFKFB3 erano in gran parte intatte. Questo suggerisce un aumento della degradazione della proteina PFKFB3 piuttosto che un problema nella sua produzione. In cellule precursori neuronali derivati da pazienti e in diversi modelli animali che imitano la patologia di TDP-43 è emerso lo stesso schema: TDP-43 mal localizzato, PFKFB3 ridotto, attività di PNKP più debole e più danni al DNA nei geni attivi.
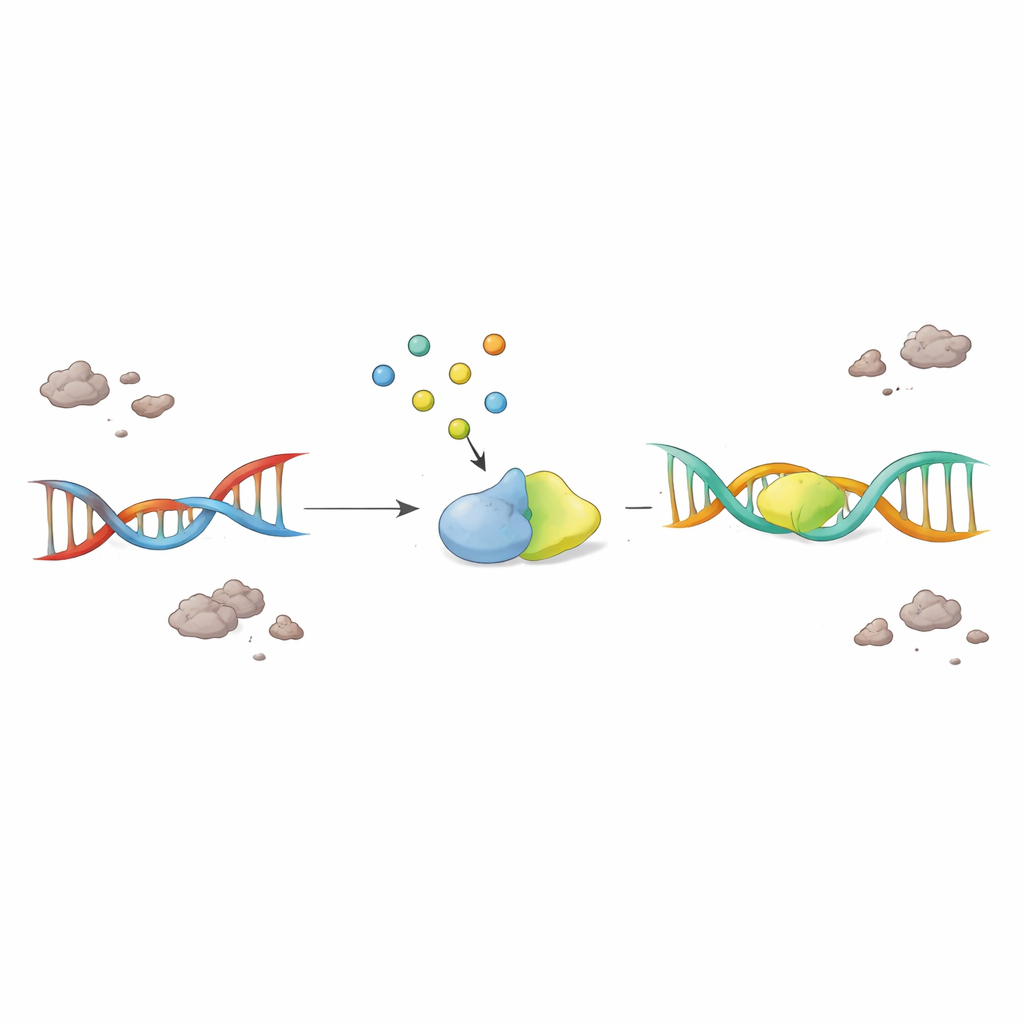
Potenziare una piccola molecola per salvare i neuroni malati
In modo cruciale, gli scienziati hanno testato se semplicemente reintegrare F2,6BP potesse riattivare il sistema di riparazione bloccato. Quando F2,6BP è stata aggiunta a estratti nucleari provenienti da cervelli di pazienti con SLA e FTD, l’attività di PNKP è ricomparsa in modo dose-dipendente, mentre molecole zuccherine strettamente correlate non avevano effetto. In cellule derivate da pazienti portatrici di mutazioni di TDP-43 legate alla malattia, F2,6BP ha sia ripristinato l’attività di PNKP sia ridotto la quantità di TDP-43 anormale e aggregata nel citoplasma. In un modello murino con patologia di TDP-43, F2,6BP ha nuovamente ravvivato la funzione di PNKP. Più sorprendente, in un modello di moscerino della frutta che esprimeva la TDP-43 umana mutata nei motoneuroni, il trattamento orale con F2,6BP ha migliorato l’abilità di arrampicata delle mosche e ridotto il danno al DNA in geni grandi e molto attivi sensibili alla rottura.
Cosa potrebbe significare per i trattamenti futuri
Nel loro insieme, questi risultati delineano uno scenario in cui la patologia di TDP-43 fa deragliare un asse critico che collega il metabolismo energetico alla riparazione del DNA: PFKFB3 produce F2,6BP, F2,6BP alimenta PNKP e PNKP protegge i geni attivi dall’accumulo di rotture. Quando questo asse fallisce, i neuroni perdono gradualmente stabilità genomica e funzionalità. Reintegrando F2,6BP, gli autori sono riusciti a riattivare l’attività di PNKP, riparare il DNA in modo più efficiente e alleviare i problemi di movimento in un organismo vivente. Pur restando molto lavoro da fare per tradurre questi risultati in terapie umane e per mettere a punto gli effetti metabolici, lo studio suggerisce che indirizzare con attenzione questa via metabolico–riparativa potrebbe aprire una nuova strada basata sui meccanismi per trattare SLA, FTD e possibilmente altri disturbi caratterizzati dall’accumulo di proteina TDP-43.
Citazione: Chakraborty, A., Mitra, J., Malojirao, V.H. et al. Fructose-2,6-bisphosphate restores TDP-43 pathology-driven genome repair deficiency in motor neuron diseases. Commun Biol 9, 563 (2026). https://doi.org/10.1038/s42003-026-09787-5
Parole chiave: ALS, TDP-43, Riparazione del DNA, Neurodegenerazione, Metabolismo cellulare