Clear Sky Science · sv
Fruktos-2,6-bisfosfat återställer TDP-43-patologiorsakad brist i genomreparation vid motorneuronsjukdomar
Varför brutet DNA spelar roll i nervceller
Amyotrofisk lateralskleros (ALS) och frontotemporal demens (FTD) är förödande hjärnsjukdomar som berövar människor rörlighet, tal och minne. Under lång tid har forskare känt till att ett protein kallat TDP-43 klumpar ihop sig på fel plats inne i nervceller vid dessa tillstånd, men hur detta leder till neuroners död har förblivit oklart. Denna studie avslöjar en oväntad koppling mellan energimetabolism och cellens förmåga att reparera brutet DNA, och visar att en liten sockerrelaterad molekyl till viss del kan vända både DNAskador och rörelsestörningar i sjukdomsmodeller.
En trafikstockning i cellens reparationsverkstad
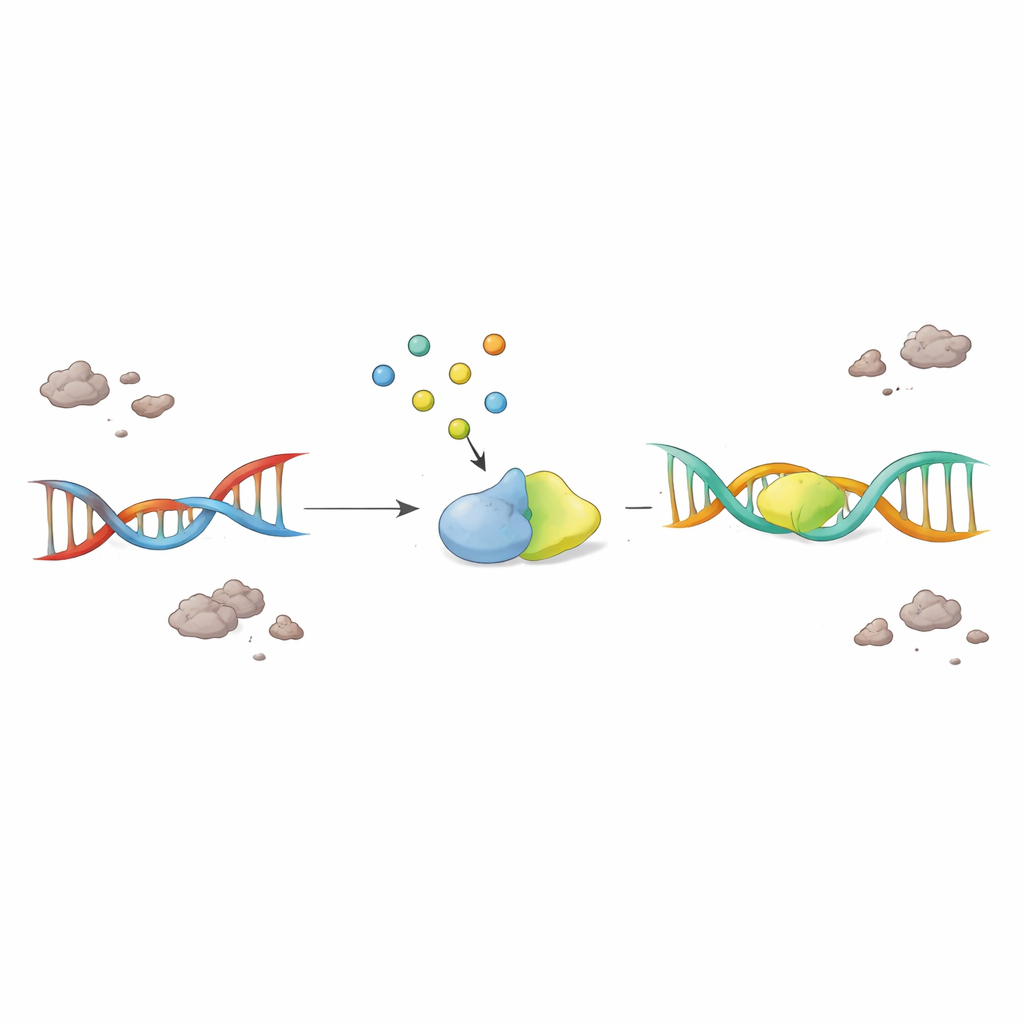
Nervceller utsätts ständigt för DNA-brott, särskilt i gener som aktivt läses av för att hålla cellen igång. Normalt hjälper ett enzym kallat PNKP till att städa upp de lösa ändarna vid brutet DNA så att reparationen kan slutföras. Författarna fann att i hjärnvävnad från personer med ALS och FTD var PNKP:s aktivitet starkt nedsatt, även om enzymet själv fortfarande var närvarande. Samtidigt fanns tydliga tecken på kvarstående DNAskador i aktiva gener, vilket tyder på att reparationsmaskineriet stod still precis där det behövdes som mest för att bevara neuronernas funktion.
Hur ett felplacerat protein stör DNA-reparation
TDP-43 finns normalt i cellkärnan, där DNA lagras, och hjälper till att samordna reparationen av farliga dubbelsträngsbrott. Vid ALS och FTD läcker emellertid TDP-43 ut i cellens yttre region och bildar klibbiga aggregat. Genom att sänka TDP-43-nivåerna i odlade celler visade forskarna att denna förlust av kärn-TDP-43 är nog för att försvaga PNKP:s förmåga att bearbeta brutet DNA, trots att PNKP:s mängd inte förändras. De observerade också att ett större reparationskomplex, som normalt inkluderar TDP-43, PNKP och andra faktorer som tätar DNA-brott i aktiva gener, faller isär när TDP-43 är fellokaliserat. Resultatet är en ansamling av DNAskador specifikt i flitigt använda gener, vilket gör redan sårbara motorneuroner ännu mer ömtåliga.
En saknad länk mellan sockeranvändning och skydd av genomet
Studien spårar sedan denna reparationssvikt tillbaka till en metabolisk omkopplare. PNKP är beroende av en liten molekyl kallad fruktos-2,6-bisfosfat (F2,6BP), som produceras av enzymet PFKFB3, för att fungera effektivt. I hjärnprover från ALS- och FTD-patienter var PFKFB3-protein och nivåerna av F2,6BP markant reducerade, medan de genetiska instruktionerna för PFKFB3 i stort sett var intakta. Detta tyder på en ökad nedbrytning av PFKFB3-proteinet snarare än ett problem med dess produktion. I patienthärledda nervprekursorceller och i flera olika djurmodeller som efterliknar TDP-43-patologi framträdde samma mönster: fellokaliserat TDP-43, lägre PFKFB3, svagare PNKP-aktivitet och mer DNAskador i aktiva gener.
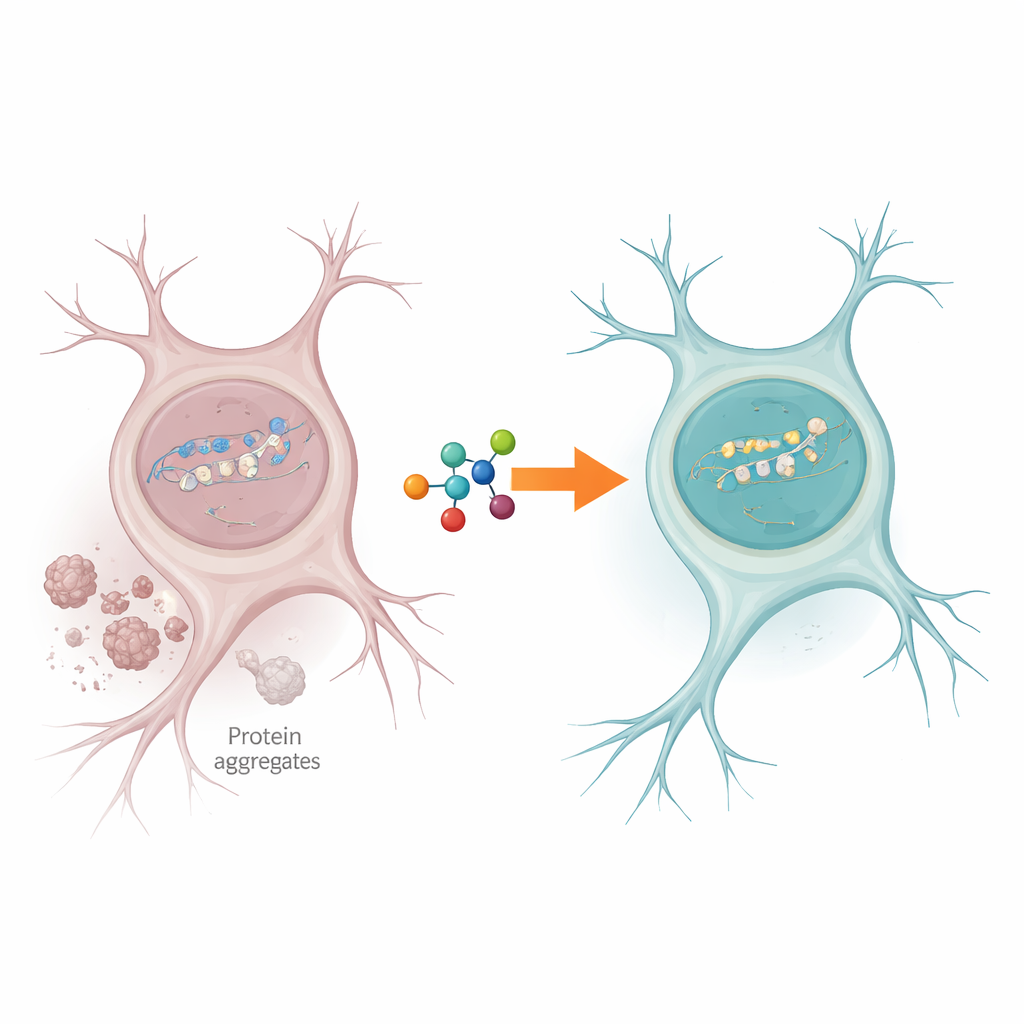
Att förstärka en liten molekyl för att rädda sjuka neuroner
Avgörande testade forskarna om man enkelt kunde återaktivera det fastnade reparationssystemet genom att tillsätta F2,6BP. När F2,6BP tillfördes nukleära extrakt från ALS- och FTD-hjärnor återhämtade sig PNKP-aktiviteten på ett dosberoende sätt, medan nära besläktade sockerarter inte hade någon effekt. I patienthärledda celler med sjukdomsassocierade TDP-43-mutationer återställde F2,6BP både PNKP-aktiviteten och minskade mängden onormalt, aggregerat TDP-43 i cytosolen. I en musmodell med TDP-43-patologi återupplivade F2,6BP återigen PNKP-funktionen. Mest anmärkningsvärt var att i en bananflugemodell som uttryckte mutant humant TDP-43 i motorneuroner förbättrade oral behandling med F2,6BP flugornas klättringsförmåga och minskade DNAskador i stora, aktivitetsintensiva gener som är känsliga för brott.
Vad detta kan innebära för framtida behandlingar
Tillsammans målar dessa fynd upp en bild där TDP-43-patologi stör en kritisk axel som kopplar energimetabolism till DNA-reparation: PFKFB3 producerar F2,6BP, F2,6BP driver PNKP, och PNKP skyddar aktiva gener från att samla på sig brott. När denna axel fallerar förlorar neuroner gradvis genomstabilitet och funktion. Genom att fylla på F2,6BP kunde författarna återuppliva PNKP-aktivitet, reparera DNA mer effektivt och lindra rörelsestörningar i en levande organism. Även om mycket arbete återstår för att översätta detta till mänskliga terapier och finjustera metaboliska effekter, tyder studien på att riktade insatser mot denna metaboliska–reparationsväg kan öppna en ny, mekanismbaserad väg för behandling av ALS, FTD och möjligen andra störningar präglade av TDP-43-ansamlingar.
Citering: Chakraborty, A., Mitra, J., Malojirao, V.H. et al. Fructose-2,6-bisphosphate restores TDP-43 pathology-driven genome repair deficiency in motor neuron diseases. Commun Biol 9, 563 (2026). https://doi.org/10.1038/s42003-026-09787-5
Nyckelord: ALS, TDP-43, DNA-reparation, neurodegeneration, cellmetabolism