Clear Sky Science · de
Fructose-2,6-bisphosphat stellt in durch TDP-43 verursachter Genomreparatur-Defizienz bei Motoneuronerkrankungen die Reparatur wieder her
Warum gebrochene DNA in Nervenzellen wichtig ist
Amyotrophe Lateralsklerose (ALS) und frontotemporale Demenz (FTD) sind verheerende Erkrankungen des Gehirns, die Menschen ihrer Bewegungsfähigkeit, Sprache und Erinnerung berauben. Seit Jahren ist bekannt, dass ein Protein namens TDP-43 in diesen Erkrankungen an falschen Stellen in Nervenzellen verklumpt, doch wie dies zum Zelltod von Neuronen führt, blieb unklar. Diese Studie zeigt eine überraschende Verbindung zwischen dem Energiestoffwechsel und der Fähigkeit der Zelle, gebrochene DNA zu reparieren, und demonstriert, dass ein kleines zuckerähnliches Molekül sowohl DNA-Schäden als auch Bewegungsstörungen in Krankheitsmodellen teilweise rückgängig machen kann.
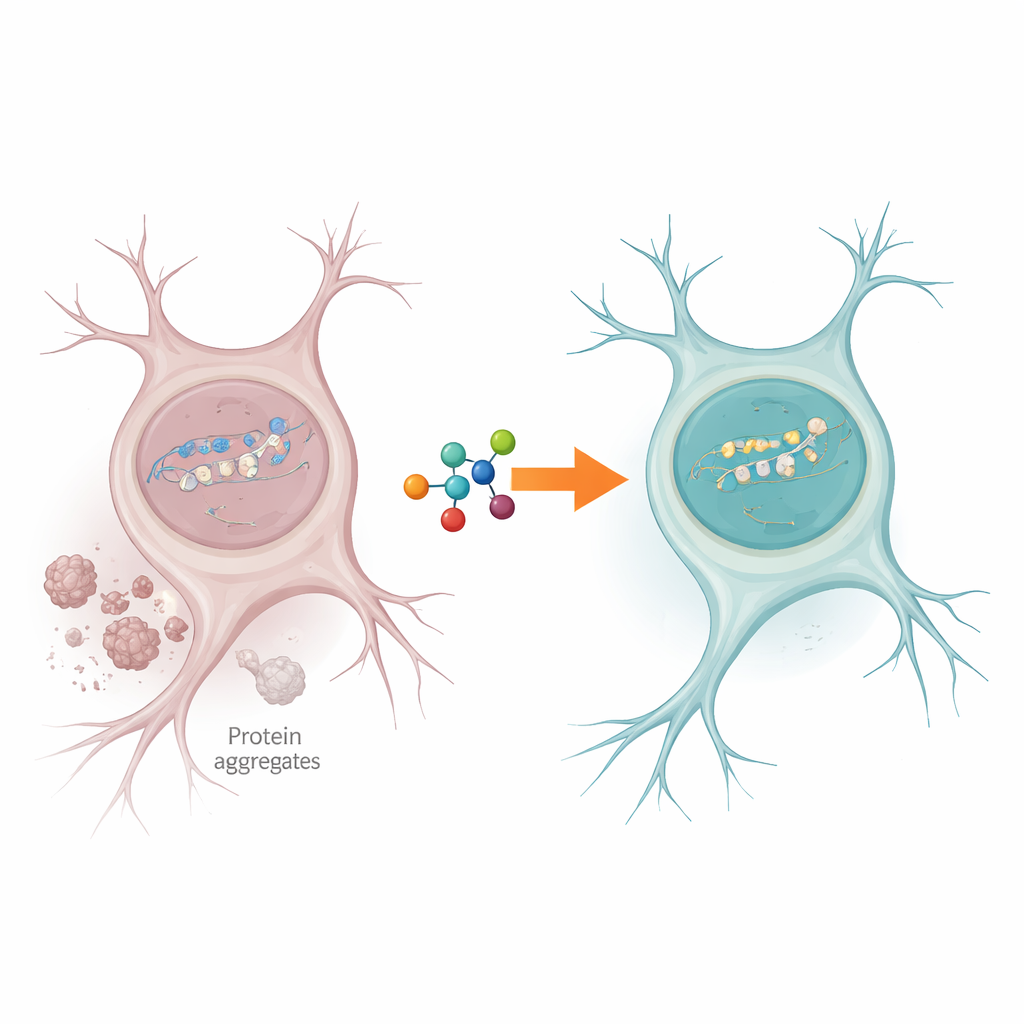
Ein Stau in der Reparaturwerkstatt der Zelle
Nervenzellen erleben ständig DNA-Brüche, besonders in Genen, die aktiv abgelesen werden, um die Zellfunktion aufrechtzuerhalten. Normalerweise hilft ein Enzym namens PNKP dabei, die offenen Enden gebrochener DNA aufzubereiten, damit die Reparatur abgeschlossen werden kann. Die Autoren fanden heraus, dass in Gehirngewebe von Menschen mit ALS und FTD die Aktivität von PNKP stark reduziert war, obwohl das Enzym selbst weiterhin vorhanden war. Gleichzeitig gab es klare Hinweise auf anhaltende DNA-Schäden in aktiven Genen, was darauf hindeutet, dass die Reparaturmaschinerie genau dort stecken blieb, wo sie am dringendsten benötigt wird, um die Funktion der Neuronen zu erhalten.
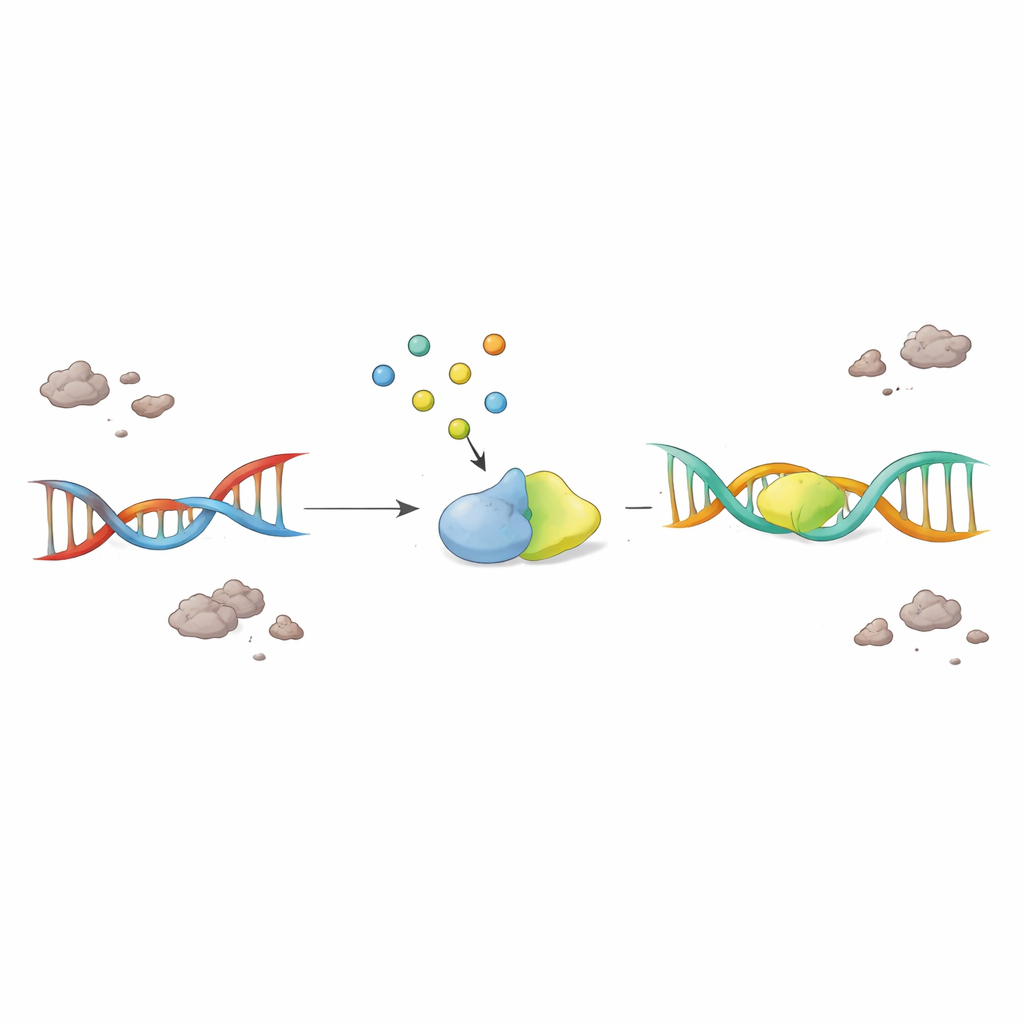
Wie ein fehlplatzierter Protein die DNA-Reparatur stört
Normalerweise befindet sich TDP-43 im Zellkern, wo die DNA lagert, und es hilft bei der Koordination der Reparatur gefährlicher Doppelstrangbrüche. Bei ALS und FTD jedoch entweicht TDP-43 ins Zytoplasma und bildet dort klebrige Aggregate. Indem die Forschenden die TDP-43-Spiegel in kultivierten Zellen senkten, zeigten sie, dass dieser Verlust von nukleärem TDP-43 ausreicht, um die Fähigkeit von PNKP, gebrochene DNA zu verarbeiten, zu schwächen, obwohl die Menge an PNKP unverändert bleibt. Sie beobachteten außerdem, dass ein größeres Reparaturkomplex, das normalerweise TDP-43, PNKP und weitere Faktoren umfasst, die aktive Gene verschließen, auseinanderfällt, wenn TDP-43 fehlplatziert ist. Das Ergebnis ist eine Anhäufung von DNA-Schäden speziell in stark genutzten Genen, wodurch ohnehin anfällige Motoneurone noch fragiler werden.
Eine fehlende Verbindung zwischen Zuckerstoffwechsel und Genomschutz
Die Studie verfolgt diesen Reparaturausfall weiter bis zu einem metabolischen Schalter. PNKP ist auf ein kleines Molekül angewiesen, das Fructose-2,6-bisphosphat (F2,6BP) heißt und vom Enzym PFKFB3 gebildet wird, um effizient funktionieren zu können. In Gehirnproben von ALS- und FTD-Patienten waren PFKFB3-Protein und F2,6BP-Spiegel deutlich reduziert, während die genetische Information für PFKFB3 weitgehend intakt war. Das deutet darauf hin, dass das PFKFB3-Protein vermehrt abgebaut wird, statt dass die Produktion gestört ist. In patientenabgeleiteten Nervenvorläuferzellen und in mehreren Tiermodellen, die die TDP-43-Pathologie nachahmen, zeigte sich dasselbe Muster: fehlplatziertes TDP-43, geringere PFKFB3-Spiegel, abgeschwächte PNKP-Aktivität und vermehrte DNA-Schäden in aktiven Genen.
Ein kleines Molekül stärken, um kranke Neurone zu retten
Entscheidend testeten die Forschenden, ob das einfache Hinzufügen von F2,6BP das feststeckende Reparatursystem wiederbeleben kann. Wurde F2,6BP zu nuklearen Extrakten aus ALS- und FTD-Gehirnen gegeben, erholte sich die PNKP-Aktivität dosisabhängig, während eng verwandte Zuckermoleküle keine Wirkung zeigten. In patientenabgeleiteten Zellen mit krankheitsassoziierten TDP-43-Mutationen stellte F2,6BP sowohl die PNKP-Aktivität wieder her als auch die Menge an abnormalem, aggregiertem TDP-43 im Zytosol verringert. In einem Mausmodell mit TDP-43-Pathologie belebte F2,6BP erneut die PNKP-Funktion. Am eindrücklichsten verbesserte in einem Fruchtfliegenmodell, das mutiertes humanes TDP-43 in Motoneuronen exprimiert, eine orale F2,6BP-Behandlung die Kletterfähigkeit der Fliegen und reduzierte DNA-Schäden in großen, aktivitätsintensiven Genen, die bruchanfällig sind.
Was das für künftige Therapien bedeuten könnte
Zusammen zeichnen diese Befunde ein Bild, in dem die TDP-43-Pathologie eine kritische Achse stört, die den Energiestoffwechsel mit der DNA-Reparatur verbindet: PFKFB3 produziert F2,6BP, F2,6BP treibt PNKP an, und PNKP schützt aktive Gene vor der Anhäufung von Brüchen. Versagt diese Achse, verlieren Neurone allmählich genomische Stabilität und Funktion. Durch die Auffüllung von F2,6BP konnten die Autoren PNKP-Aktivität wiederbeleben, die DNA-Reparatur effizienter machen und Bewegungsprobleme in einem lebenden Organismus lindern. Obwohl noch viel Arbeit nötig ist, um dies in humane Therapien zu überführen und metabolische Nebenwirkungen präzise zu steuern, legt die Studie nahe, dass eine gezielte Beeinflussung dieses Stoffwechsel–Reparatur-Wegs eine neue, mechanismusbasierte Möglichkeit zur Behandlung von ALS, FTD und möglicherweise anderen Störungen mit TDP-43-Akkumulation eröffnen könnte.
Zitation: Chakraborty, A., Mitra, J., Malojirao, V.H. et al. Fructose-2,6-bisphosphate restores TDP-43 pathology-driven genome repair deficiency in motor neuron diseases. Commun Biol 9, 563 (2026). https://doi.org/10.1038/s42003-026-09787-5
Schlüsselwörter: ALS, TDP-43, DNA-Reparatur, Neurodegeneration, Zellstoffwechsel