Clear Sky Science · fr
Le fructose-2,6-bisphosphate restaure la déficience de réparation du génome liée à la pathologie TDP-43 dans les maladies des motoneurones
Pourquoi l’ADN cassé compte dans les cellules nerveuses
La sclérose latérale amyotrophique (SLA) et la démence frontotemporale (DFT) sont des maladies cérébrales dévastatrices qui privent les personnes de la parole, du mouvement et de la mémoire. Depuis des années, les chercheurs savent qu’une protéine appelée TDP-43 s’agrège au mauvais endroit à l’intérieur des cellules nerveuses dans ces affections, mais le lien entre ces amas et la mort neuronale est resté obscure. Cette étude révèle un lien surprenant entre le métabolisme énergétique et la capacité de la cellule à réparer l’ADN brisé, et montre qu’une petite molécule liée aux sucres peut en partie inverser à la fois les dommages à l’ADN et les troubles moteurs dans des modèles de la maladie.
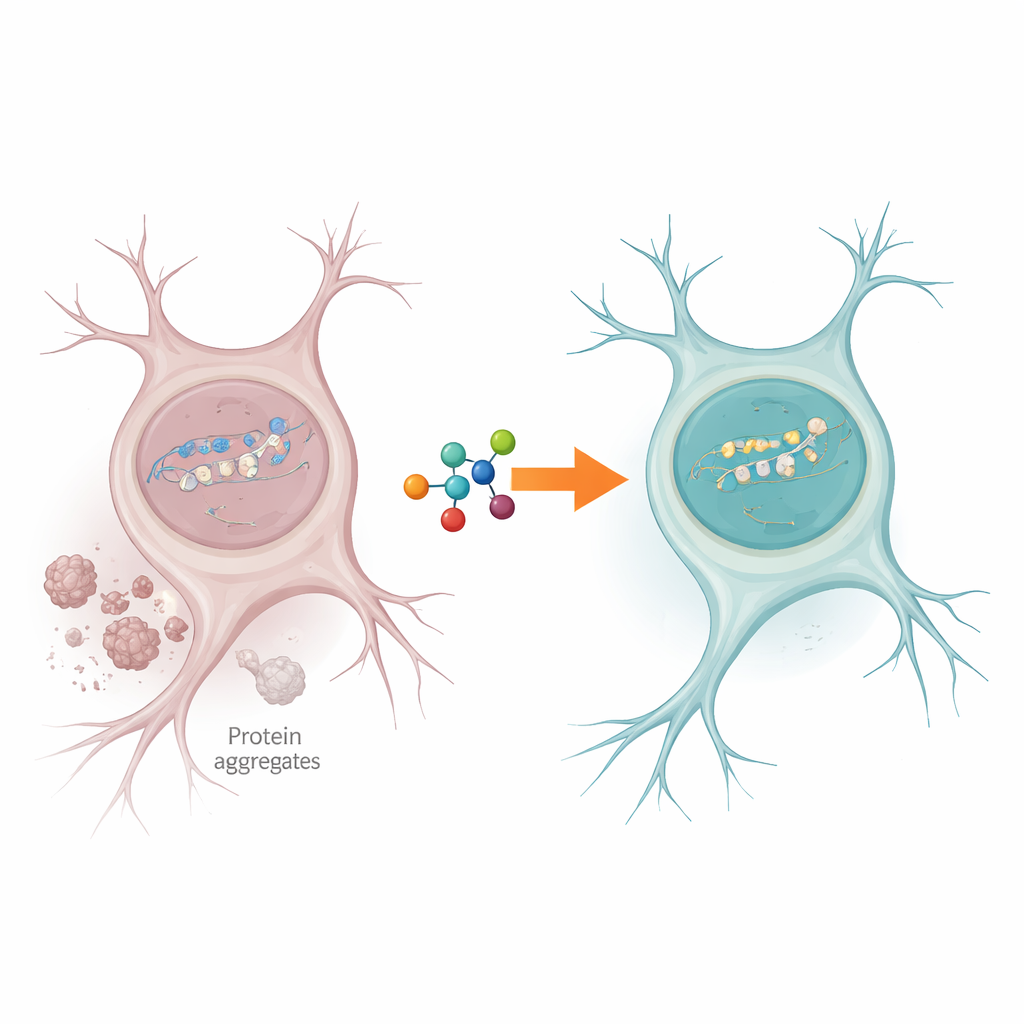
Un embouteillage dans l’atelier de réparation de la cellule
Les cellules nerveuses subissent constamment des cassures d’ADN, en particulier dans les gènes activement lus pour assurer le fonctionnement cellulaire. Normalement, une enzyme nommée PNKP aide à nettoyer les extrémités lâches de l’ADN brisé pour permettre la réparation. Les auteurs ont constaté que, dans des tissus cérébraux de personnes atteintes de SLA et de DFT, l’activité de PNKP était fortement réduite, alors que l’enzyme elle-même était encore présente. Parallèlement, il existait des preuves nettes de dommages persistants dans les gènes actifs, ce qui suggère que la machinerie de réparation était bloquée précisément là où elle est la plus nécessaire pour préserver la fonction neuronale.
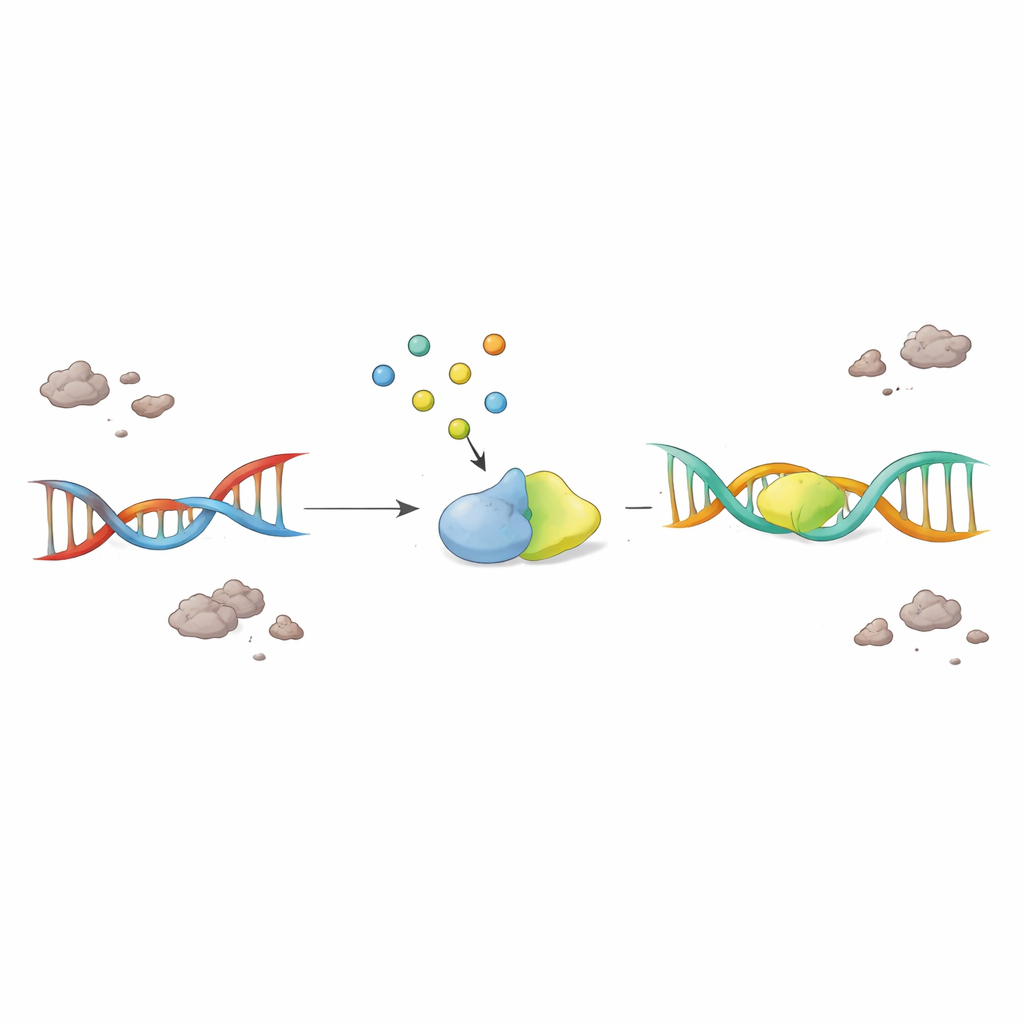
Comment une protéine mal placée perturbe la réparation de l’ADN
TDP-43 réside normalement dans le noyau, où est stocké l’ADN, et aide à coordonner la réparation des cassures double-brin dangereuses. Dans la SLA et la DFT, toutefois, TDP-43 fuit vers le cytoplasme et forme des agrégats collants. En réduisant le niveau de TDP-43 dans des cellules en culture, les chercheurs ont montré que cette perte de TDP-43 nucléaire suffit à réduire drastiquement la capacité de PNKP à traiter l’ADN brisé, même si la quantité de PNKP ne change pas. Ils ont également observé qu’une équipe de réparation plus large, qui comprend normalement TDP-43, PNKP et d’autres facteurs qui scellent les cassures dans les gènes actifs, se défait lorsque TDP-43 est mal localisé. Le résultat est une accumulation de dommages à l’ADN spécifiquement dans les gènes très sollicités, rendant des motoneurones déjà vulnérables encore plus fragiles.
Un maillon manquant entre l’utilisation du sucre et la protection du génome
L’étude ramène ensuite cette défaillance de réparation à un changement métabolique. PNKP dépend d’une petite molécule appelée fructose-2,6-bisphosphate (F2,6BP), produite par l’enzyme PFKFB3, pour fonctionner efficacement. Dans les échantillons cérébraux de SLA et de DFT, les protéines PFKFB3 et les niveaux de F2,6BP étaient nettement réduits, tandis que les instructions génétiques pour PFKFB3 restaient globalement intactes. Cela suggère une augmentation de la dégradation de la protéine PFKFB3 plutôt qu’un défaut de production. Dans des cellules précurseurs neuronales dérivées de patients et dans plusieurs modèles animaux reproduisant la pathologie TDP-43, le même schéma est apparu : TDP-43 mal localisé, PFKFB3 diminué, activité de PNKP affaiblie et plus de dommages à l’ADN dans les gènes actifs.
Augmenter une petite molécule pour sauver des neurones malades
De manière cruciale, les scientifiques ont testé si l’ajout simple de F2,6BP pouvait relancer le système de réparation bloqué. Lorsqu’on a fourni du F2,6BP à des extraits nucléaires issus de cerveaux de patients atteints de SLA et de DFT, l’activité de PNKP a rebondi de façon dépendante de la dose, tandis que des molécules sucrées étroitement apparentées n’avaient aucun effet. Dans des cellules dérivées de patients portant des mutations de TDP-43 liées à la maladie, le F2,6BP a à la fois restauré l’activité de PNKP et réduit la quantité de TDP-43 anormale et agrégée dans le cytosol. Dans un modèle murin présentant la pathologie TDP-43, le F2,6BP a de nouveau ravivé la fonction de PNKP. Plus frappant encore, dans un modèle de drosophile exprimant la TDP-43 humaine mutante dans les motoneurones, un traitement oral au F2,6BP a amélioré la capacité d’escalade des mouches et réduit les dommages à l’ADN dans de grands gènes très actifs et sensibles aux cassures.
Ce que cela pourrait signifier pour les traitements futurs
Ensemble, ces résultats dessinent un scénario dans lequel la pathologie TDP-43 perturbe un axe critique reliant le métabolisme énergétique à la réparation de l’ADN : PFKFB3 produit le F2,6BP, le F2,6BP alimente PNKP, et PNKP protège les gènes actifs contre l’accumulation de cassures. Lorsque cet axe faillit, les neurones perdent progressivement la stabilité et la fonction génomiques. En reconstituant le F2,6BP, les auteurs ont pu relancer l’activité de PNKP, améliorer la réparation de l’ADN et atténuer les problèmes moteurs chez un organisme vivant. Bien qu’il reste beaucoup à faire pour traduire cela en thérapies humaines et pour ajuster les effets métaboliques, l’étude suggère que cibler soigneusement cette voie métabolique–réparation pourrait ouvrir une nouvelle voie thérapeutique fondée sur les mécanismes pour traiter la SLA, la DFT et possiblement d’autres troubles marqués par l’accumulation de la protéine TDP-43.
Citation: Chakraborty, A., Mitra, J., Malojirao, V.H. et al. Fructose-2,6-bisphosphate restores TDP-43 pathology-driven genome repair deficiency in motor neuron diseases. Commun Biol 9, 563 (2026). https://doi.org/10.1038/s42003-026-09787-5
Mots-clés: SLA, TDP-43, Réparation de l’ADN, Neurodégénérescence, métabolisme cellulaire