Clear Sky Science · nl
Fructose-2,6-bisfosfaat herstelt door TDP-43-pathologie veroorzaakte tekortkomingen in DNA-reparatie bij motorneuronziekten
Waarom gebroken DNA belangrijk is in zenuwcellen
Amyotrofische laterale sclerose (ALS) en frontotemporale dementie (FTD) zijn verwoestende hersenziekten die mensen hun beweging, spraak en geheugen ontnemen. Jarenlang wisten wetenschappers dat een eiwit genaamd TDP-43 op de verkeerde plaats in zenuwcellen klontert bij deze aandoeningen, maar hoe dat leidt tot het afsterven van neuronen bleef vaag. Deze studie onthult een verrassende koppeling tussen energiemetabolisme en het vermogen van de cel om gebroken DNA te repareren, en laat zien dat een klein suikergerelateerd molecuul deels zowel DNA-schade als bewegingsproblemen in ziektemodellen kan omkeren.
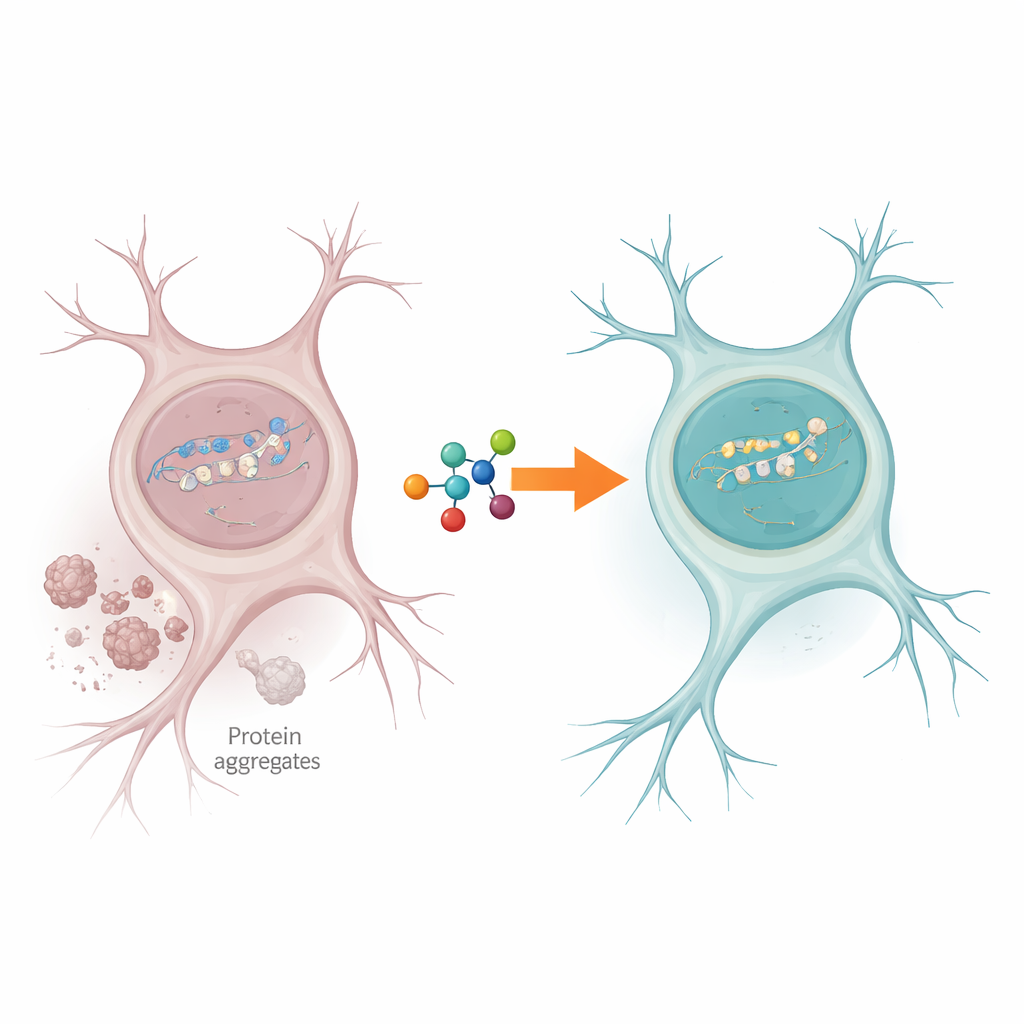
Een verkeersopstopping in de reparatiewerkplaats van de cel
Zenuwcellen ondervinden voortdurend DNA-breuken, vooral in genen die actief worden afgelezen om de cel te laten functioneren. Normaal helpt een enzym genaamd PNKP de losse eindjes van gebroken DNA op te schonen zodat reparatie kan worden voltooid. De auteurs vonden dat in hersenweefsel van mensen met ALS en FTD de activiteit van PNKP sterk verminderd was, zelfs hoewel het enzym zelf nog aanwezig was. Tegelijkertijd waren er duidelijke aanwijzingen voor aanhoudende DNA-schade in actieve genen, wat suggereert dat de reparatiemachine vastloopt juist daar waar die het meest nodig is om de functie van neuronen te bewaren.
Hoe een verplaatste eiwit DNA-reparatie verstoort
TDP-43 bevindt zich normaal in de kern, waar DNA wordt opgeslagen, en helpt bij het coördineren van de reparatie van gevaarlijke dubbelstrengs breuken. Bij ALS en FTD lekt TDP-43 echter uit naar het buitenste deel van de cel en vormt plakkerige aggregaten. Door TDP-43-niveaus te verlagen in gekweekte cellen toonden de onderzoekers aan dat dit verlies van kern-TDP-43 voldoende is om PNKP’s vermogen om gebroken DNA te verwerken te verlammen, ook al verandert de hoeveelheid PNKP niet. Ze observeerden ook dat een groter reparatieteam, dat normaal TDP-43, PNKP en andere factoren omvat die DNA-breuken in actieve genen afsluiten, uiteenvalt wanneer TDP-43 verkeerd is gelokaliseerd. Het resultaat is een ophoping van DNA-schade specifiek in intens gebruikte genen, waardoor reeds kwetsbare motorneuronen nog fragieler worden.
Een ontbrekende schakel tussen suikergebruik en genoombescherming
De studie traceert dit reparatiefalen vervolgens terug naar een metabole schakel. PNKP is afhankelijk van een klein molecuul genaamd fructose-2,6-bisfosfaat (F2,6BP), geproduceerd door het enzym PFKFB3, om efficiënt te functioneren. In ALS- en FTD-hersenmonsters waren PFKFB3-eiwit en F2,6BP-niveaus duidelijk verlaagd, terwijl de genetische instructies voor PFKFB3 grotendeels intact bleven. Dit wijst op verhoogde afbraak van het PFKFB3-eiwit in plaats van een probleem met de productie. In patiënt-afgeleide voorlopercellen van zenuwcellen en in meerdere diermodellen die TDP-43-pathologie nabootsen, kwam hetzelfde patroon naar voren: verkeerd gelokaliseerd TDP-43, lagere PFKFB3, verminderde PNKP-activiteit en meer DNA-schade in actieve genen.
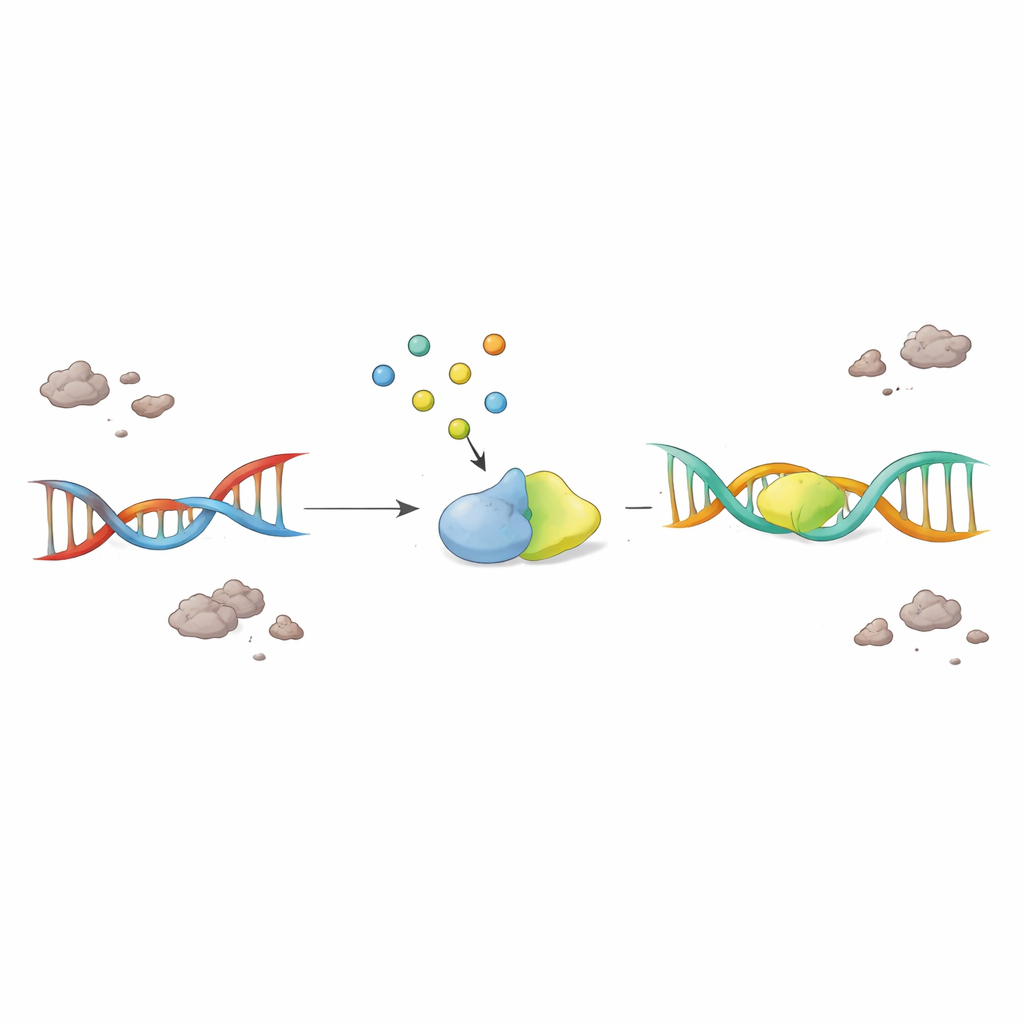
Het stimuleren van een klein molecuul om zieke neuronen te redden
Crisisachtig testten de wetenschappers of het simpelweg teruggeven van F2,6BP het vastgelopen reparatiesysteem weer tot leven kon wekken. Wanneer F2,6BP aan kernextracten uit ALS- en FTD-hersenen werd toegevoegd, herstelde de PNKP-activiteit in een dosisafhankelijke manier, terwijl sterk verwante suikermoleculen geen effect hadden. In patiënt-afgeleide cellen met ziektegerelateerde TDP-43-mutaties herstelde F2,6BP zowel de PNKP-activiteit als verminderde het de hoeveelheid abnormaal, geaggregeerd TDP-43 in het cytosol. In een muismodel met TDP-43-pathologie herleefde F2,6BP opnieuw de PNKP-functie. Het meest opvallend was dat in een fruitvliegmodel dat mutant menselijk TDP-43 in motorneuronen tot expressie brengt, orale F2,6BP-behandeling het klimvermogen van de vliegen verbeterde en DNA-schade verminderde in grote, actief gebruikte genen die gevoelig zijn voor breuk.
Wat dit kan betekenen voor toekomstige behandelingen
Samen schetsen deze bevindingen een beeld waarin TDP-43-pathologie een cruciale as ontspoort die energiemetabolisme koppelt aan DNA-reparatie: PFKFB3 produceert F2,6BP, F2,6BP voedt PNKP, en PNKP beschermt actieve genen tegen het ophopen van breuken. Wanneer deze as faalt, verliezen neuronen geleidelijk hun genomische stabiliteit en functie. Door F2,6BP aan te vullen konden de auteurs PNKP-activiteit nieuw leven inblazen, DNA efficiënter repareren en bewegingsproblemen in een levend organisme verzachten. Hoewel er nog veel werk nodig is om dit naar menselijke therapieën te vertalen en de metabole effecten fijn af te stemmen, suggereert de studie dat gerichte interventie in deze metabole–reparatie-route een nieuwe, op het mechanisme gebaseerde benadering kan openen voor de behandeling van ALS, FTD en mogelijk andere aandoeningen gekenmerkt door TDP-43-eiwitophoping.
Bronvermelding: Chakraborty, A., Mitra, J., Malojirao, V.H. et al. Fructose-2,6-bisphosphate restores TDP-43 pathology-driven genome repair deficiency in motor neuron diseases. Commun Biol 9, 563 (2026). https://doi.org/10.1038/s42003-026-09787-5
Trefwoorden: ALS, TDP-43, DNA-reparatie, neurodegeneratie, celmetabolisme