Clear Sky Science · tr
FNIP2-SERCA2b eksenini hedeflemek Ataksi Telanjiektazideki metabolik ve mitokondriyal kusurları iyileştirir
Hücrenin DNA koruyucusu enerji yönetimini de sağladığında
Ataksi telanjiektazi, hareket bozuklukları, bağışıklık zayıflığı ve artmış kanser riskiyle tanınan nadir bir genetik hastalıktır. Bu çalışma, bu hastalardaki eksik ATM proteininin yalnızca DNA’yı korumakla kalmayıp aynı zamanda hücrelerin şekeri enerji için nasıl kullandığını da ana hatlarıyla yönettiğini gösteriyor. ATM kaybolduğunda hücreler sadece DNA hasarıyla mücadele etmekle kalmıyor—glukozu da yanlış yönetiyor, fazla glikojenle tıkanıyor ve enerji santralleri olan mitokondrileri yakıtsız bırakıyor. Çalışma, bu enerji dengesini kısmen geri getirebilecek yeni bir moleküler anahtarın varlığına işaret ediyor.
Nadir Bir Hastalıkta Enerji Sorunları
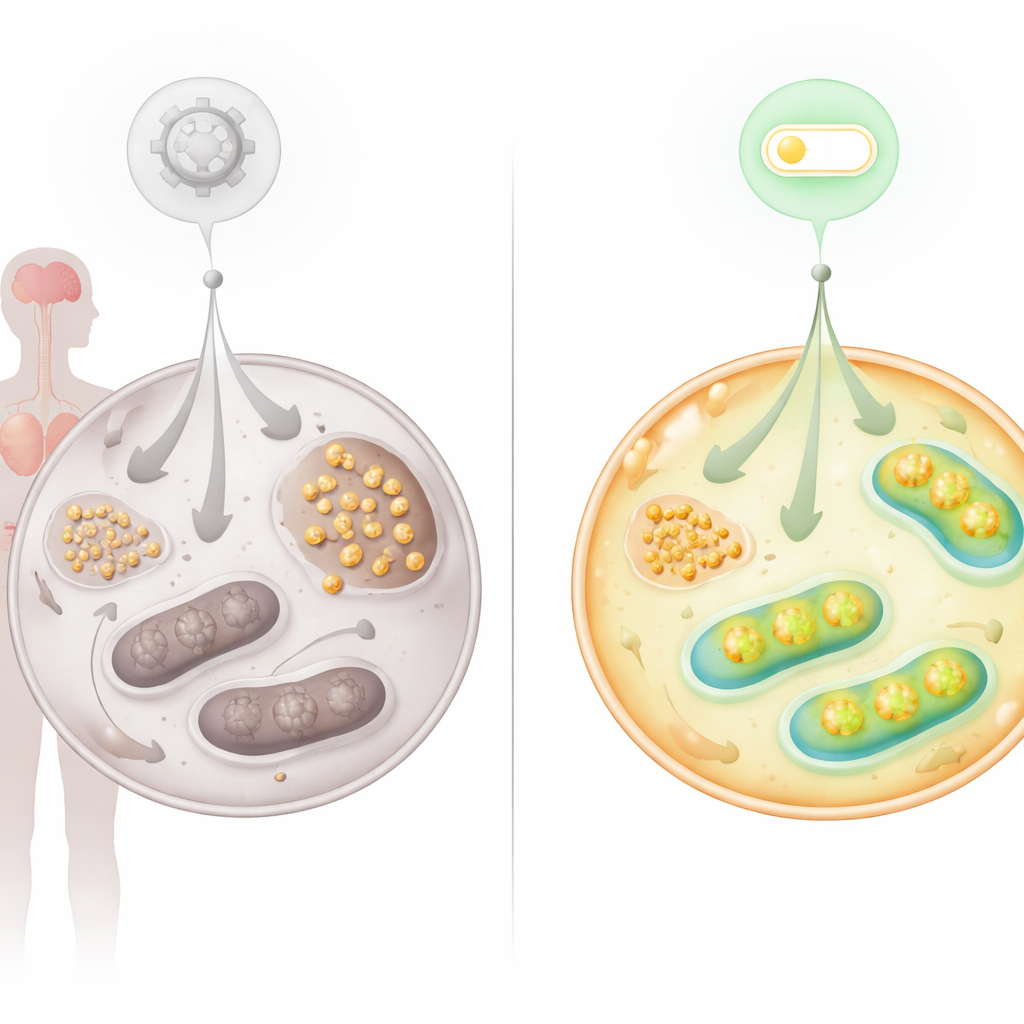
Araştırmacılar, ataksi telanjiektazi hastalarından ve sağlıklı gönüllülerden alınan deri hücreleriyle başladılar. Geniş metabolit profillemesi ve oksijen tüketimi ölçümleri kullanarak, AT hücrelerinin kronik oksidatif stres halinde yaşadığını ve yakıtı kötü yaktığını buldular. Şeker yıkımının (glikoliz) ana adımları ve mitokondrideki sonraki enerji üreten döngü her ikisi de yavaşlamıştı. Sonuç olarak, AT hücreleri alternatif yakıt arıyormuş gibi normalden daha fazla protein ve nükleik asit yıkımı yaptı. Bu kusurlar tek bir bilinen enzim eksikliğinin tekrarı değildi; ATM kaybının daha geniş, sistem düzeyinde bir metabolik bozulmaya yol açtığını göstermektedir.
Şekerin Çıkmaz Bir Depoya Dönüşmesi
Canlı hücrelerde işaretli glukoz izlenerek, AT hücrelerinde gelen şekerin daha azının ana enerji yollarına aktığı ortaya çıktı. Bunun yerine glikojen metabolizmasıyla ilişkili yıkım ürünleri birikti. Mikroskopi, dallanmış bir glukoz depolama formu olan glikojenin AT fibroblastlarında, bu fibroblastlardan yeniden programlanan kök hücrelerde ve kalp ile beyincik dahil AT hastalarının dokularında yoğun bir şekilde biriktiğini doğruladı. Hareket kontrolü için kritik olan beyinciğin sinir hücreleri anormal glikojen birikintileri ve glikojen yapan enzimin artmış düzeylerini gösterdi. Bu desen, mitokondriler yakıtı verimli bir şekilde işleyemediğinde hücrelerin fazla glukozu depoya yönlendirdiğini ve bunun da beyin ve kalp gibi hassas organlarda ek strese yol açabileceğini öne sürer.
Yeni Bir Anahtar Bulmak: FNIP2–SERCA2b Ekseni
Metabolik sorunlar mitokondri kusurlarına güçlü bir şekilde işaret ettiğinden ekip, mitokondriyal aktiviteyi açıp kapatan proteinlere baktı. Besin ve redoks stresine yanıt vermeye yardımcı olan FNIP1’in bir akrabası olan FNIP2’ye odaklandılar. AT hücrelerinde FNIP2’nin susturulması glikojen birikimini dramatik şekilde azalttı ve hücrelerin zaman içinde koloniler oluşturma yeteneğini geri getirerek bu hücreleri genellikle etkileyen erken yaşlanmayı geciktirdi. Ayrıntılı bioenerjetik testler, AT hücrelerinde FNIP2’nin bloke edilmesinin glikolizi artırdığını, mitokondriyal solunumu ve ATP üretimini yükselttiğini gösterdi; böylece glukoz kullanımı sağlıklı hücrelere aşırı düzeltme yapmadan normale daha yakın hale geldi.
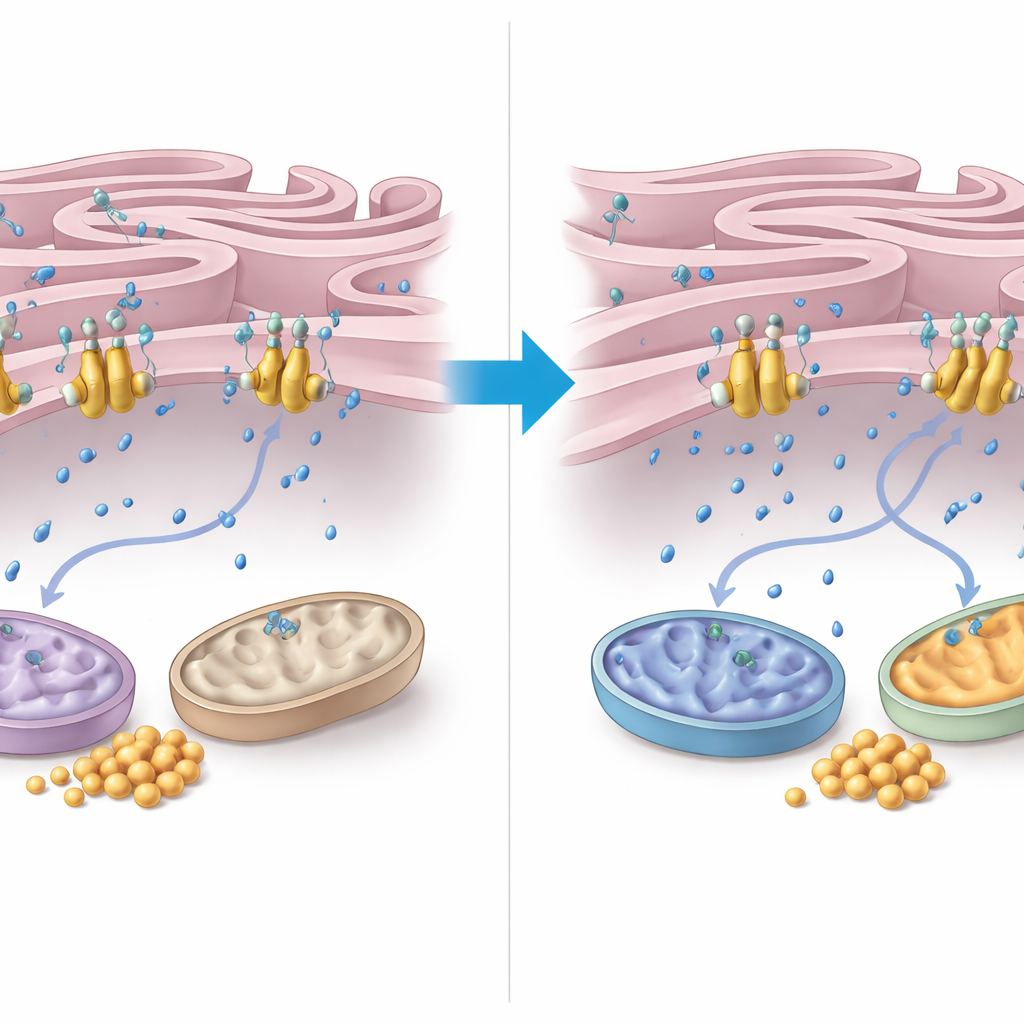
Kalsiyumun Kurtarıcı Sinyalini Nasıl Aktardığı
FNIP2’nin bu kontrolü nasıl uyguladığını anlamak için yazarlar endoplazmik retikulum (ER) ile mitokondri arasındaki kalsiyum taşınmasına odaklandı. FNIP2’nin ER zarındaki bir kalsiyum pompası olan SERCA2b’ye sıkı şekilde bağlandığını keşfettiler. Hücre tabanlı deneylerde FNIP2 çıkarıldığında ER’nin kalsiyum alımı keskin şekilde azaldı; bu, daha fazla kalsiyumun çevredeki sitoplazmada kaldığı anlamına geliyordu. Bu ekstra kalsiyum mitokondriyal enzimleri ve solunumu uyarabilir. Elektron mikroskopisi, AT hücrelerinin bozulmuş mitokondrilere ve ER ile mitokondri arasında olağanüstü derecede sıkı, geniş temas bölgelerine sahip olduğunu gösterdi—bunlar oksidatif stresle ilişkili özelliklerdir. Çarpıcı olarak, FNIP2 baskılanması yalnızca mitokondri şeklini normale döndürmekle kalmadı, aynı zamanda daha tipik ER–mitokondri temas desenlerini geri getirerek daha sağlıklı bir enerji yönetimiyle uyumlu sonuçlar verdi.
Hastalar İçin Anlamı
Bir araya getirildiğinde bulgular, ataksi telanjiektaziyi sadece bir DNA onarım bozukluğu olarak değil aynı zamanda hücresel yakıtın yanlış yönetildiği bir hastalık olarak yeniden tanımlıyor. ATM olmadan glukoz enerji için az kullanılıyor, mitokondriler düşük performans gösteriyor ve glikojen kritik organlarda hücreleri tıkıyor. FNIP2’yi kısmen azaltarak—dolayısıyla SERCA2b kalsiyum pompasının uyarımını hafifleterek—hücreler mitokondriyal aktiviteyi canlandırmak, daha fazla glukoz yakmak ve glikojen birikimini sınırlamak için sitoplazmik kalsiyumu yeterince yükseltebilir. Bu çalışma hücreler ve doku örneklerinde yapılmış olsa da FNIP2–SERCA2b yolunu, metabolizmayı ve hücre hayatta kalımını iyileştirmeyi amaçlayan potansiyel terapilere yönelik umut verici bir hedef olarak tanımlamaktadır.
Atıf: Vinciguerra, M., El Kharef, C., Bruhn, C. et al. Targeting the FNIP2-SERCA2b axis improves metabolic and mitochondrial defects in Ataxia Telangiectasia. Cell Death Dis 17, 290 (2026). https://doi.org/10.1038/s41419-026-08507-5
Anahtar kelimeler: ataksi telanjiektazi, hücresel metabolizma, mitokondri, kalsiyum sinyalleşmesi, glikojen birikimi