Clear Sky Science · sv
Att rikta FNIP2–SERCA2b-axeln förbättrar metabola och mitokondriella defekter vid ataxi-telangiektasi
När cellens DNA-vakt också styr energihanteringen
Ataxi-telangiektasi är främst känt som en sällsynt genetisk sjukdom som orsakar rörelsestörningar, nedsatt immunförsvar och ökad cancerrisk. Denna studie visar att förutom att skydda vårt DNA fungerar den saknade ATM-proteinet hos dessa patienter också som en central regissör för hur celler använder socker som energi. När ATM förloras kämpar cellerna inte bara med DNA-skador — de hanterar också glukos felaktigt, fylls av överskottligt glykogen och svälter sina kraftverk, mitokondrierna. Arbetet pekar på en ny molekylär brytare som kan växlas för att delvis återställa denna energibalans.
Energiproblem i en sällsynt sjukdom
Forskarna började med hudceller tagna från personer med ataxi-telangiektasi och från friska frivilliga. Med bred metabolitprofilering och mätningar av syreförbrukning fann de att AT-celler lever i ett tillstånd av kronisk oxidativ stress och förbrukar bränsle ineffektivt. Centrala steg i sockerbrytningen (glykolys) och den efterföljande energigenererande cykeln i mitokondrierna gick båda långsamt. Som ett resultat bröt AT-celler ner proteiner och nukleinsyror mer än normalt, som om de spanade efter alternativa bränslen. Dessa defekter var inte helt enkelt en upprepning av ett känt enzymbristtillstånd, vilket tyder på att förlusten av ATM orsakar ett bredare, systemnivåfelskick i metabolismen.
När socker blir en återvändsgränd i form av förråd
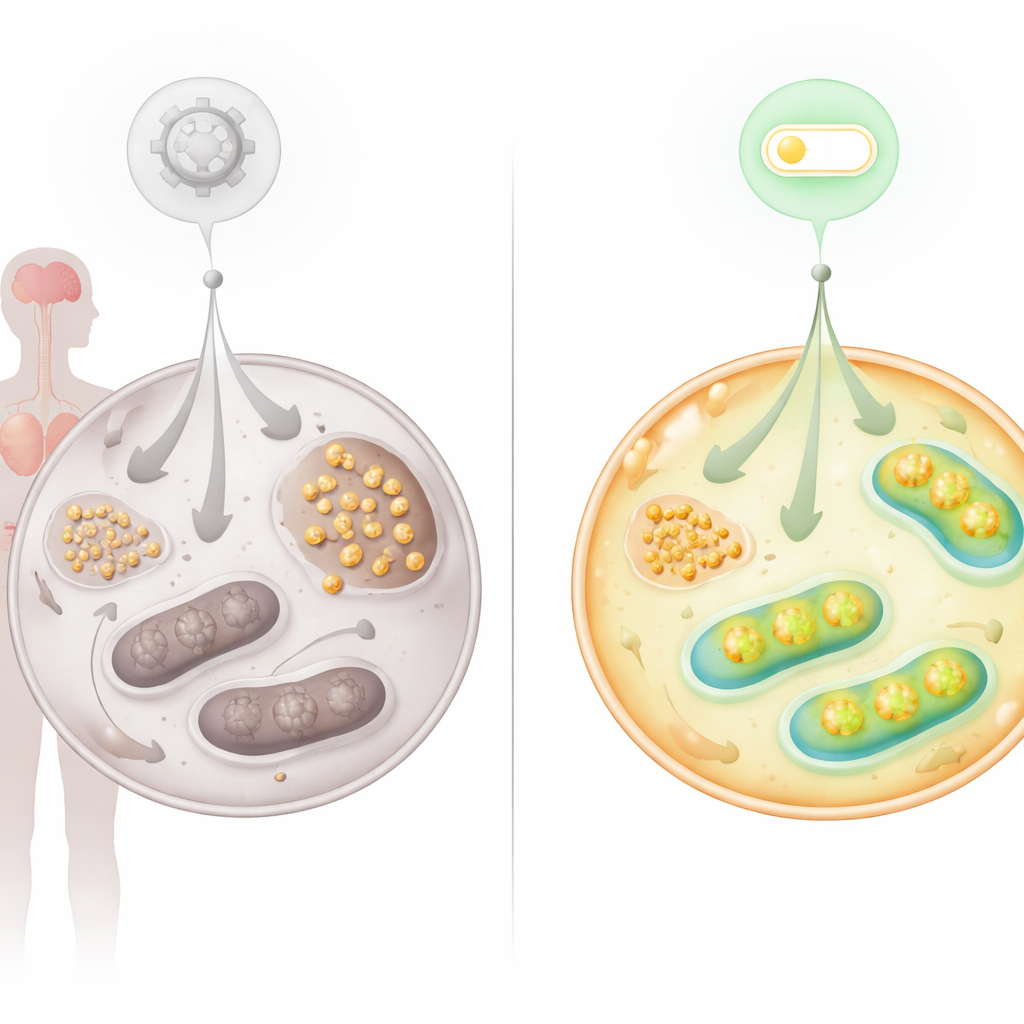
Spårning av märkt glukos i levande celler visade att en mindre andel av det inkommande sockret flöt genom huvudvägarna för energi i AT-celler. Istället ackumulerades nedbrytningsprodukter kopplade till glykogenmetabolism. Mikroskopi bekräftade att glykogen — en förgrenad lagringsform av glukos — byggdes upp i hög grad i AT-fibroblaster, i stamceller omprogrammerade från dessa fibroblaster och i vävnader från AT-patienter, inklusive hjärta och lillhjärna. Nervceller i lillhjärnan, viktiga för rörelsekontroll, visade onormala glykogendepåer och förhöjda nivåer av enzymet som bildar glykogen. Detta mönster tyder på att när mitokondrier inte effektivt kan bearbeta bränsle, styr cellerna överskottet av glukos in i lagring, vilket i sin tur kan belasta känsliga organ som hjärna och hjärta.
Att hitta en ny brytare: FNIP2–SERCA2b-axeln
Eftersom de metabola problemen tydde starkt på felande mitokondrier, undersökte teamet proteiner som är kända för att skruva upp eller ner mitokondriell aktivitet. De fokuserade på FNIP2, en släkting till FNIP1, som hjälper celler att svara på närings- och redoxstress. Att tysta FNIP2 i AT-celler minskade dramatiskt glykogenuppbyggnaden och återställde deras förmåga att bilda kolonier över tid, vilket försenade den för tidiga senescensen som vanligtvis drabbar dessa celler. Detaljerade bioenergetiska tester visade att blockering av FNIP2 i AT-celler förbättrade glykolysen och ökade mitokondriell andning och ATP-produktion, vilket förde glukosanvändningen närmare normal utan att överkorrigera i friska celler.
Hur kalcium förmedlar räddningssignalen
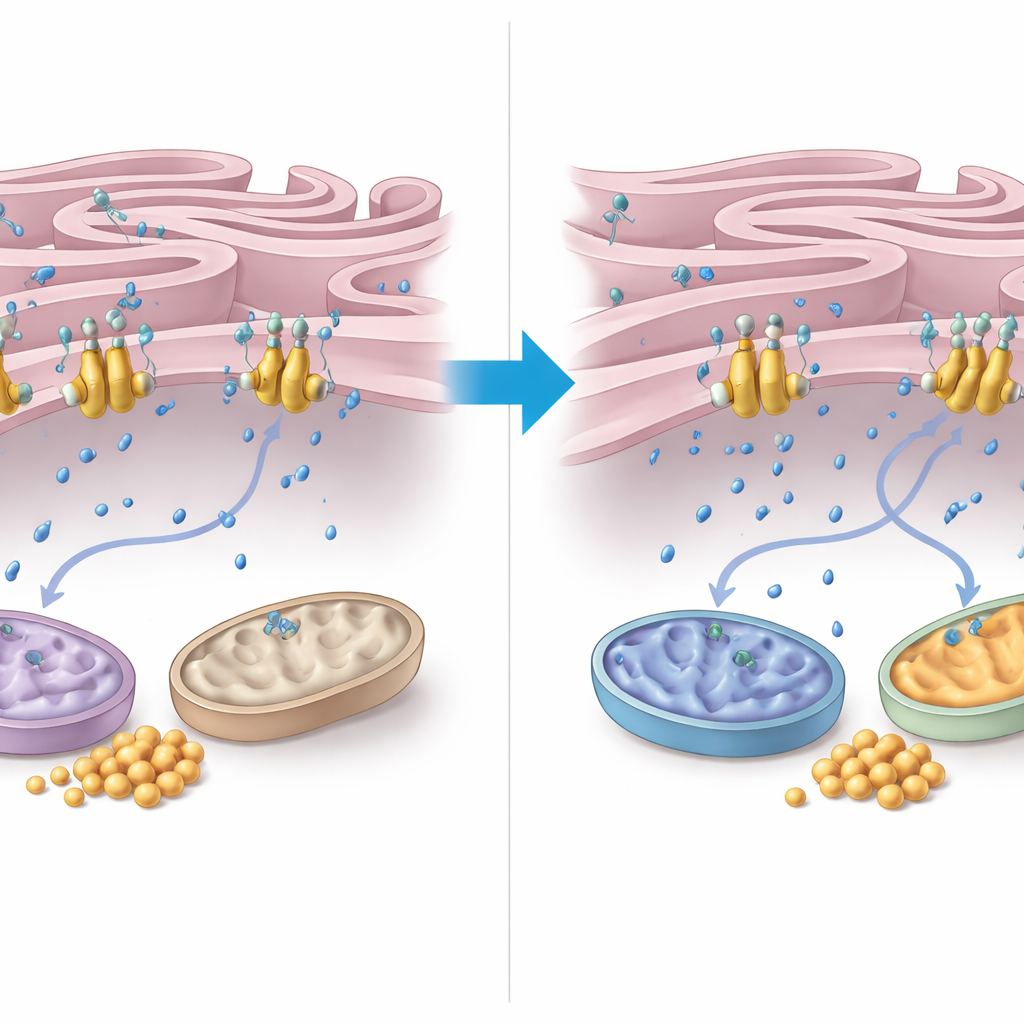
För att förstå hur FNIP2 utövar denna kontroll fördjupade författarna sig i kalciumhanteringen mellan endoplasmatiskt retikulum (ER) och mitokondrier. De upptäckte att FNIP2 binder starkt till en kalciumpump i ER-membranet kallad SERCA2b. I cellbaserade tester minskade borttagning av FNIP2 kraftigt ER:s upptag av kalcium, vilket innebar att mer kalcium blev kvar i omgivande cytoplasma. Detta extra kalcium kan stimulera mitokondriella enzymer och andning. Elektronmikroskopi visade att AT-celler har förvrängda mitokondrier och ovanligt täta, vidsträckta kontaktytor mellan ER och mitokondrier — drag kopplade till oxidativ stress. Slående nog normaliserade FNIP2-suppression inte bara mitokondriernas form utan återställde också mer typiska ER–mitokondrie-kontaktmönster, i linje med en hälsosammare energihantering.
Vad detta betyder för patienter
Tillsammans omformulerar fynden ataxi-telangiektasi som inte bara en DNA-reparationsstörning utan också en sjukdom av felhanterat cellulärt bränsle. Utan ATM används glukos i för liten utsträckning för energi, mitokondrierna presterar sämre och glykogen täpper igen celler i kritiska organ. Genom att dämpa FNIP2 — och därigenom lätta dess stimulans av SERCA2b-kalciumpumpen — kan celler höja cytoplasmatiskt kalcium precis tillräckligt för att återuppliva mitokondriell aktivitet, förbränna mer glukos och begränsa glykogenansamling. Även om detta arbete gjordes i celler och vävnadsprover identifierar det FNIP2–SERCA2b-vägen som ett lovande mål för terapier som syftar till att förbättra metabolism och cellsöverlevnad hos personer som lever med detta komplexa tillstånd.
Citering: Vinciguerra, M., El Kharef, C., Bruhn, C. et al. Targeting the FNIP2-SERCA2b axis improves metabolic and mitochondrial defects in Ataxia Telangiectasia. Cell Death Dis 17, 290 (2026). https://doi.org/10.1038/s41419-026-08507-5
Nyckelord: ataxi-telangiektasi, cellmetabolism, mitokondrier, kalciumsignalering, glykogenansamling