Clear Sky Science · en
Targeting the FNIP2-SERCA2b axis improves metabolic and mitochondrial defects in Ataxia Telangiectasia
When the Cell’s DNA Guardian Also Manages Energy
Ataxia telangiectasia is best known as a rare genetic disease that causes movement problems, immune weakness, and cancer risk. This study shows that, beyond guarding our DNA, the missing ATM protein in these patients also acts as a master manager of how cells use sugar for energy. When ATM is lost, cells not only struggle with DNA damage—they also mishandle glucose, clogging up with excess glycogen and starving their power plants, the mitochondria. The work points to a new molecular switch that can be flipped to partly restore this energy balance.
Energy Trouble in a Rare Disease
The researchers began with skin cells taken from people with ataxia telangiectasia and from healthy volunteers. Using broad metabolite profiling and oxygen-consumption measurements, they found that AT cells live in a state of chronic oxidative stress and burn fuel poorly. Key steps of sugar breakdown (glycolysis) and the downstream energy-generating cycle in mitochondria were both sluggish. As a result, AT cells broke down proteins and nucleic acids more than normal, as if they were scavenging alternative fuels. These defects were not simply a replay of a single known enzyme deficiency, indicating that ATM loss causes a broader, system-level metabolic failure.
When Sugar Turns into a Dead-End Store
Tracing labeled glucose through living cells revealed that less of the incoming sugar flowed through the main energy pathways in AT cells. Instead, breakdown products linked to glycogen metabolism accumulated. Microscopy confirmed that glycogen—a branched storage form of glucose—built up heavily in AT fibroblasts, in stem cells reprogrammed from those fibroblasts, and in tissues from AT patients, including heart and cerebellum. Nerve cells in the cerebellum, crucial for movement control, showed abnormal glycogen deposits and heightened levels of the enzyme that makes glycogen. This pattern suggests that when mitochondria cannot efficiently process fuel, cells shunt excess glucose into storage, which may in turn stress vulnerable organs such as brain and heart.
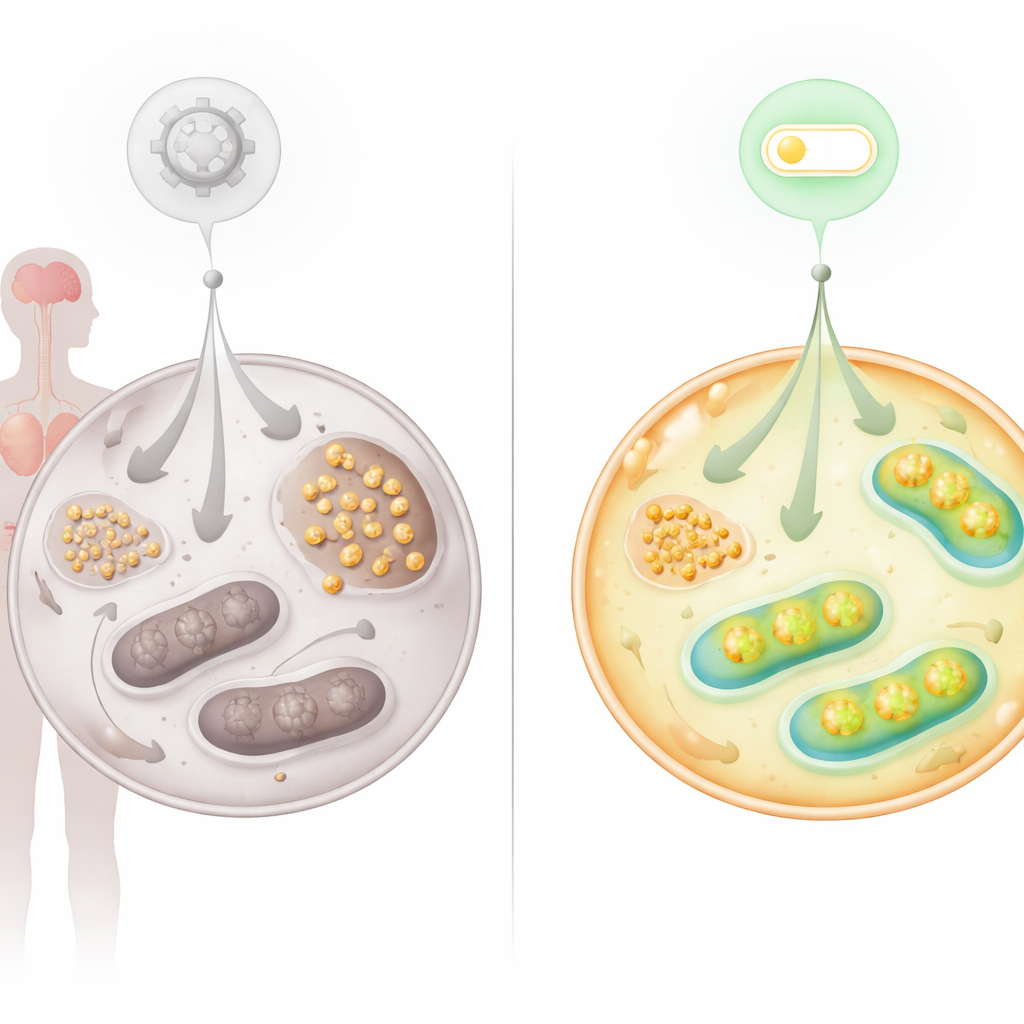
Finding a New Switch: The FNIP2–SERCA2b Axis
Because the metabolic problems pointed strongly to faulty mitochondria, the team looked at proteins known to dial mitochondrial activity up or down. They focused on FNIP2, a cousin of FNIP1, which helps cells respond to nutrient and redox stress. Silencing FNIP2 in AT cells dramatically reduced glycogen buildup and restored their ability to form colonies over time, delaying the premature senescence that usually plagues these cells. Detailed bioenergetic tests showed that blocking FNIP2 in AT cells enhanced glycolysis and increased mitochondrial respiration and ATP production, bringing glucose usage closer to normal without overcorrecting it in healthy cells.
How Calcium Relays the Rescue Signal
To understand how FNIP2 exerts this control, the authors zoomed in on calcium handling between the endoplasmic reticulum (ER) and mitochondria. They discovered that FNIP2 binds tightly to a calcium pump in the ER membrane called SERCA2b. In cell-based assays, removing FNIP2 sharply reduced the ER’s uptake of calcium, meaning more calcium remained in the surrounding cytoplasm. This extra calcium can stimulate mitochondrial enzymes and respiration. Electron microscopy revealed that AT cells have distorted mitochondria and unusually tight, extensive contact zones between ER and mitochondria—features linked to oxidative stress. Strikingly, FNIP2 suppression not only normalized mitochondrial shape but also restored more typical ER–mitochondria contact patterns, consistent with healthier energy management.
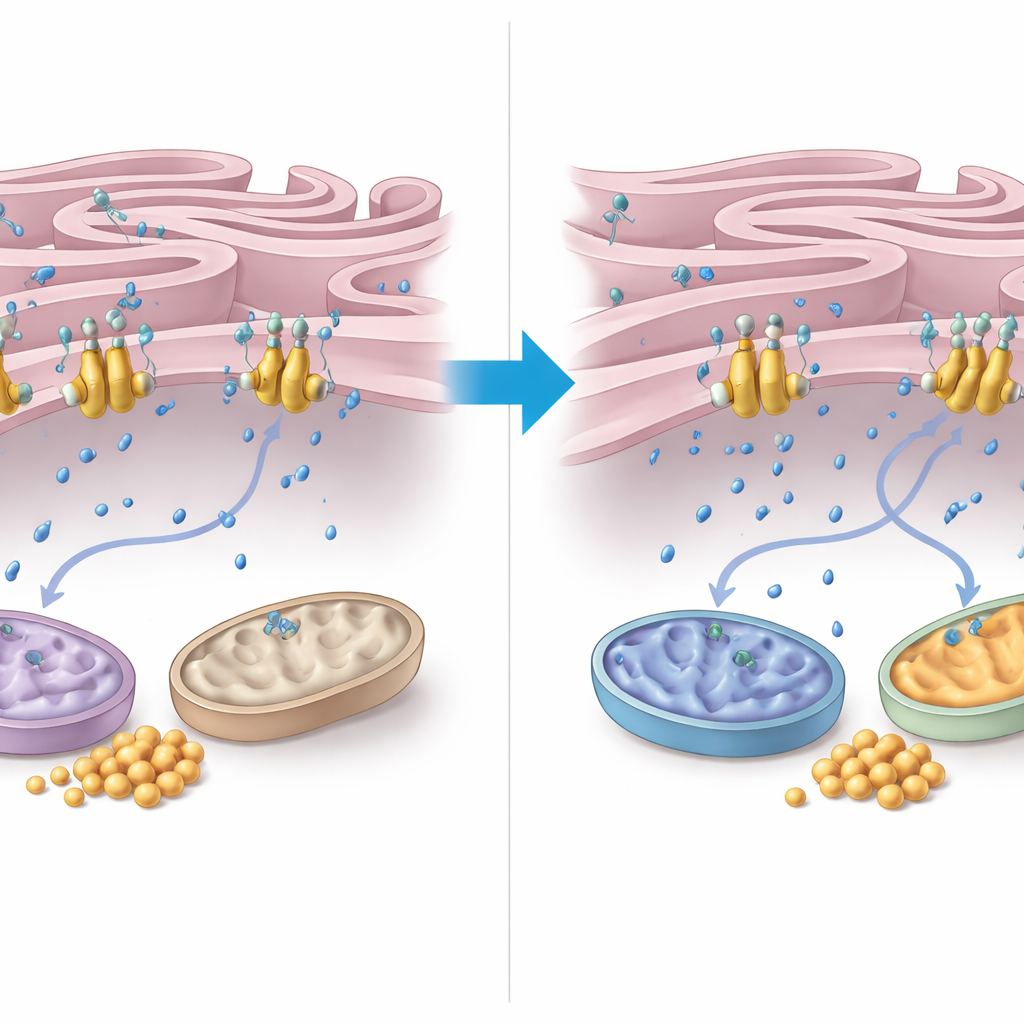
What This Means for Patients
Taken together, the findings recast ataxia telangiectasia as not just a DNA-repair disorder but also a disease of mismanaged cellular fuel. Without ATM, glucose is underused for energy, mitochondria underperform, and glycogen clogs cells in critical organs. By dialing down FNIP2—and thereby easing its stimulation of the SERCA2b calcium pump—cells can raise cytoplasmic calcium just enough to revive mitochondrial activity, burn more glucose, and limit glycogen buildup. While this work was done in cells and tissue samples, it identifies the FNIP2–SERCA2b pathway as a promising target for therapies aimed at improving metabolism and cell survival in people living with this complex condition.
Citation: Vinciguerra, M., El Kharef, C., Bruhn, C. et al. Targeting the FNIP2-SERCA2b axis improves metabolic and mitochondrial defects in Ataxia Telangiectasia. Cell Death Dis 17, 290 (2026). https://doi.org/10.1038/s41419-026-08507-5
Keywords: ataxia telangiectasia, cellular metabolism, mitochondria, calcium signaling, glycogen accumulation