Clear Sky Science · fr
Cibler l’axe FNIP2–SERCA2b améliore les défauts métaboliques et mitochondriaux dans l’ataxie télangiectasie
Quand le gardien de l’ADN de la cellule gère aussi l’énergie
L’ataxie télangiectasie est surtout connue comme une maladie génétique rare provoquant des troubles du mouvement, une fragilité immunitaire et un risque de cancer. Cette étude montre que, au-delà de la protection de notre ADN, la protéine ATM manquante chez ces patients joue aussi un rôle central dans la gestion de l’utilisation du sucre par les cellules pour produire de l’énergie. En l’absence d’ATM, les cellules ne rencontrent pas seulement des problèmes de réparation de l’ADN : elles malmènent aussi le glucose, s’encombrant d’un excès de glycogène et affamant leurs centrales énergétiques, les mitochondries. Ce travail met en lumière un nouveau commutateur moléculaire qui peut être actionné pour restaurer en partie cet équilibre énergétique.
Problèmes d’énergie dans une maladie rare
Les chercheurs ont commencé par des cellules cutanées prélevées chez des personnes atteintes d’ataxie télangiectasie et chez des volontaires sains. Par un profilage large des métabolites et des mesures de consommation d’oxygène, ils ont constaté que les cellules AT vivent dans un état de stress oxydatif chronique et brûlent mal leur carburant. Des étapes clés de la dégradation du sucre (glycolyse) et le cycle énergétique en aval dans les mitochondries étaient tous deux ralentis. En conséquence, les cellules AT dégradaient davantage de protéines et d’acides nucléiques que la normale, comme si elles cherchaient des combustibles alternatifs. Ces défauts ne se réduisaient pas à la simple reprise d’une déficience enzymatique connue, ce qui indique que la perte d’ATM provoque une défaillance métabolique plus large, à l’échelle du système.
Quand le sucre se transforme en impasse de stockage
Le suivi du glucose marqué dans des cellules vivantes a révélé qu’une moindre part du sucre entrant circulait via les principales voies énergétiques dans les cellules AT. À la place, des produits de dégradation liés au métabolisme du glycogène s’accumulaient. La microscopie a confirmé que le glycogène — une forme ramifiée de stockage du glucose — s’accumulait fortement dans les fibroblastes AT, dans les cellules souches reprogrammées à partir de ces fibroblastes, et dans les tissus de patients AT, notamment le cœur et le cervelet. Les neurones du cervelet, cruciaux pour le contrôle du mouvement, présentaient des dépôts de glycogène anormaux et des niveaux élevés de l’enzyme qui synthétise le glycogène. Ce tableau suggère que lorsque les mitochondries ne peuvent pas traiter efficacement le carburant, les cellules détournent l’excès de glucose vers le stockage, ce qui peut à son tour stresser des organes vulnérables comme le cerveau et le cœur.
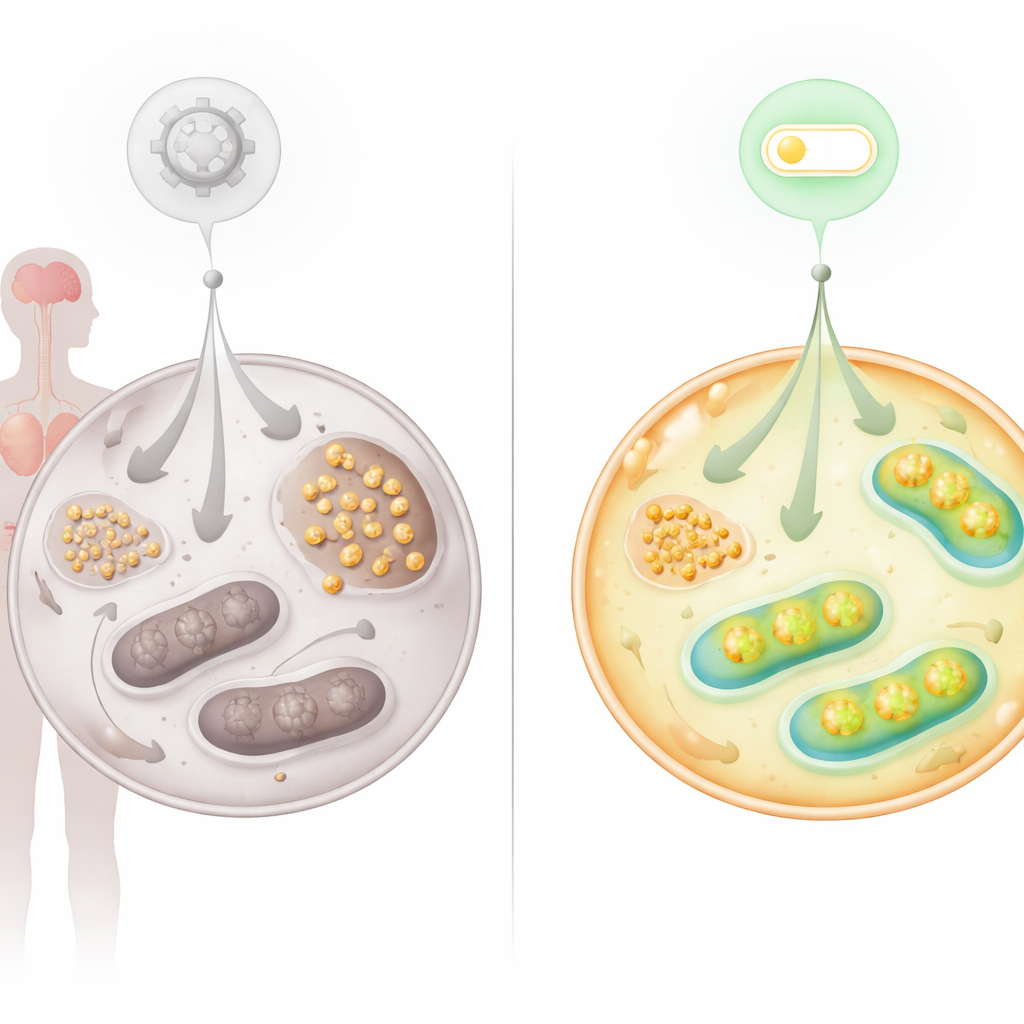
Trouver un nouveau commutateur : l’axe FNIP2–SERCA2b
Parce que les problèmes métaboliques indiquaient fortement des mitochondries déficientes, l’équipe s’est intéressée aux protéines connues pour régler l’activité mitochondriale. Ils ont focalisé sur FNIP2, un homologue de FNIP1, qui aide les cellules à répondre au stress nutritif et redox. L’inhibition de FNIP2 dans les cellules AT a réduit de façon spectaculaire l’accumulation de glycogène et a restauré leur capacité à former des colonies au fil du temps, retardant la sénescence prématurée qui affecte habituellement ces cellules. Des tests bioénergétiques détaillés ont montré que bloquer FNIP2 dans les cellules AT améliorait la glycolyse et augmentait la respiration mitochondriale et la production d’ATP, rapprochant l’utilisation du glucose d’un état plus normal sans surcorriger les cellules saines.
Comment le calcium relaie le signal de sauvetage
Pour comprendre comment FNIP2 exerce ce contrôle, les auteurs ont examiné la gestion du calcium entre le réticulum endoplasmique (RE) et les mitochondries. Ils ont découvert que FNIP2 se lie fortement à une pompe à calcium de la membrane du RE appelée SERCA2b. Dans des essais cellulaires, la suppression de FNIP2 réduisait nettement l’absorption du calcium par le RE, ce qui laissait davantage de calcium dans le cytoplasme environnant. Ce calcium supplémentaire peut stimuler des enzymes mitochondriales et la respiration. La microscopie électronique a révélé que les cellules AT ont des mitochondries déformées et des zones de contact RE–mitochondries inhabituellement serrées et étendues — des caractéristiques liées au stress oxydatif. Fait saisissant, la suppression de FNIP2 non seulement normalisait la forme mitochondriale mais rétablissait aussi des schémas de contact RE–mitochondries plus typiques, compatibles avec une gestion énergétique plus saine.
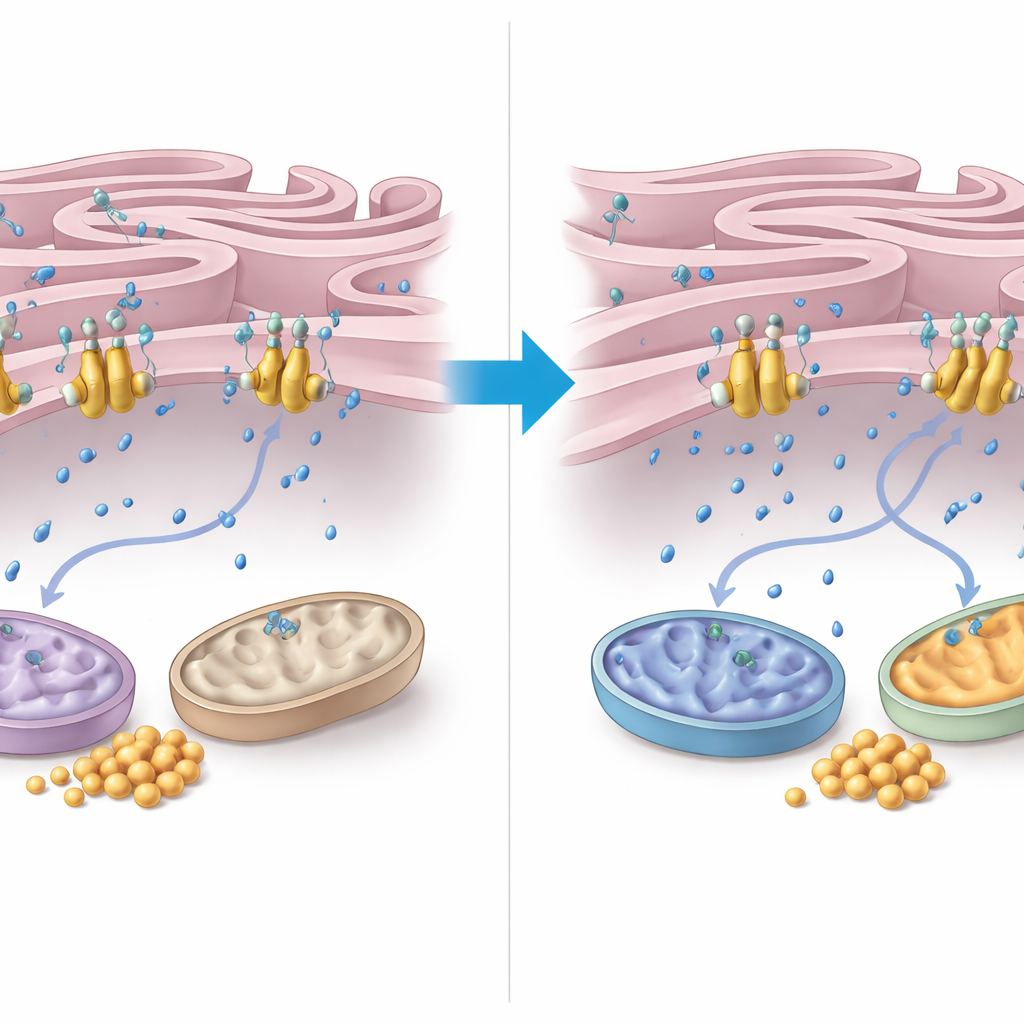
Ce que cela signifie pour les patients
Pris ensemble, ces résultats reconsidèrent l’ataxie télangiectasie non seulement comme un trouble de la réparation de l’ADN mais aussi comme une maladie de mauvaise gestion du carburant cellulaire. En l’absence d’ATM, le glucose est sous-utilisé pour produire de l’énergie, les mitochondries sont moins performantes et le glycogène encombre les cellules dans des organes critiques. En réduisant l’activité de FNIP2 — et en atténuant ainsi sa stimulation de la pompe calcique SERCA2b — les cellules peuvent augmenter le calcium cytoplasmique juste suffisamment pour ranimer l’activité mitochondriale, brûler plus de glucose et limiter l’accumulation de glycogène. Bien que ce travail ait été réalisé dans des cellules et des échantillons de tissus, il identifie la voie FNIP2–SERCA2b comme une cible prometteuse pour des thérapies visant à améliorer le métabolisme et la survie cellulaire chez les personnes vivant avec cette affection complexe.
Citation: Vinciguerra, M., El Kharef, C., Bruhn, C. et al. Targeting the FNIP2-SERCA2b axis improves metabolic and mitochondrial defects in Ataxia Telangiectasia. Cell Death Dis 17, 290 (2026). https://doi.org/10.1038/s41419-026-08507-5
Mots-clés: ataxie télangiectasie, métabolisme cellulaire, mitochondries, signalisation calcique, accumulation de glycogène