Clear Sky Science · es
Apuntar al eje FNIP2-SERCA2b mejora los defectos metabólicos y mitocondriales en la Ataxia Telangiectasia
Cuando el guardián del ADN de la célula también gestiona la energía
La ataxia telangiectasia es más conocida como una enfermedad genética rara que provoca problemas de movimiento, debilidad inmunitaria y riesgo de cáncer. Este estudio muestra que, además de proteger nuestro ADN, la proteína ATM que falta en estos pacientes actúa también como un gestor maestro de cómo las células usan el azúcar para obtener energía. Cuando se pierde ATM, las células no solo tienen problemas con el daño del ADN: también manejan mal la glucosa, acumulando glucógeno en exceso y privando de combustible a sus centrales energéticas, las mitocondrias. El trabajo señala un nuevo interruptor molecular que puede activarse para restaurar parcialmente este equilibrio energético.
Problemas energéticos en una enfermedad rara
Los investigadores comenzaron con células de la piel tomadas de personas con ataxia telangiectasia y de voluntarios sanos. Mediante perfiles amplios de metabolitos y mediciones del consumo de oxígeno, hallaron que las células AT viven en un estado de estrés oxidativo crónico y queman combustible de forma deficiente. Pasos clave de la degradación de azúcares (glucólisis) y el ciclo energético mitocondrial aguas abajo estaban retrasados. Como resultado, las células AT descomponían proteínas y ácidos nucleicos más de lo normal, como si fueran a la caza de combustibles alternativos. Estos defectos no se debieron simplemente a la repetición de una única deficiencia enzimática conocida, lo que indica que la pérdida de ATM provoca un fallo metabólico de mayor alcance, a nivel de sistema.
Cuando el azúcar se convierte en un almacenamiento sin salida
Rastrear glucosa marcada a través de células vivas reveló que menos del azúcar entrante fluía por las vías energéticas principales en las células AT. En su lugar, se acumulaban productos de degradación asociados al metabolismo del glucógeno. La microscopía confirmó que el glucógeno —una forma ramificada de almacenamiento de glucosa— se acumulaba de forma intensa en fibroblastos AT, en células madre reprogramadas a partir de esos fibroblastos y en tejidos de pacientes AT, incluidos corazón y cerebelo. Las células nerviosas del cerebelo, cruciales para el control del movimiento, mostraban depósitos anormales de glucógeno y niveles elevados de la enzima que sintetiza glucógeno. Este patrón sugiere que cuando las mitocondrias no pueden procesar combustible de manera eficiente, las células derivan el exceso de glucosa hacia el almacenamiento, lo que a su vez puede estresar órganos vulnerables como el cerebro y el corazón.
Encontrar un nuevo interruptor: el eje FNIP2–SERCA2b
Dado que los problemas metabólicos apuntaban con fuerza a mitocondrias defectuosas, el equipo examinó proteínas conocidas por regular la actividad mitocondrial. Se centraron en FNIP2, un pariente de FNIP1, que ayuda a las células a responder al estrés nutritivo y redox. Silenciar FNIP2 en células AT redujo drásticamente la acumulación de glucógeno y restauró su capacidad de formar colonias con el tiempo, retrasando la senescencia prematura que suele afectar a estas células. Pruebas bioenergéticas detalladas mostraron que bloquear FNIP2 en células AT mejoró la glucólisis e incrementó la respiración mitocondrial y la producción de ATP, acercando el uso de glucosa a la normalidad sin sobrecorregir en células sanas.
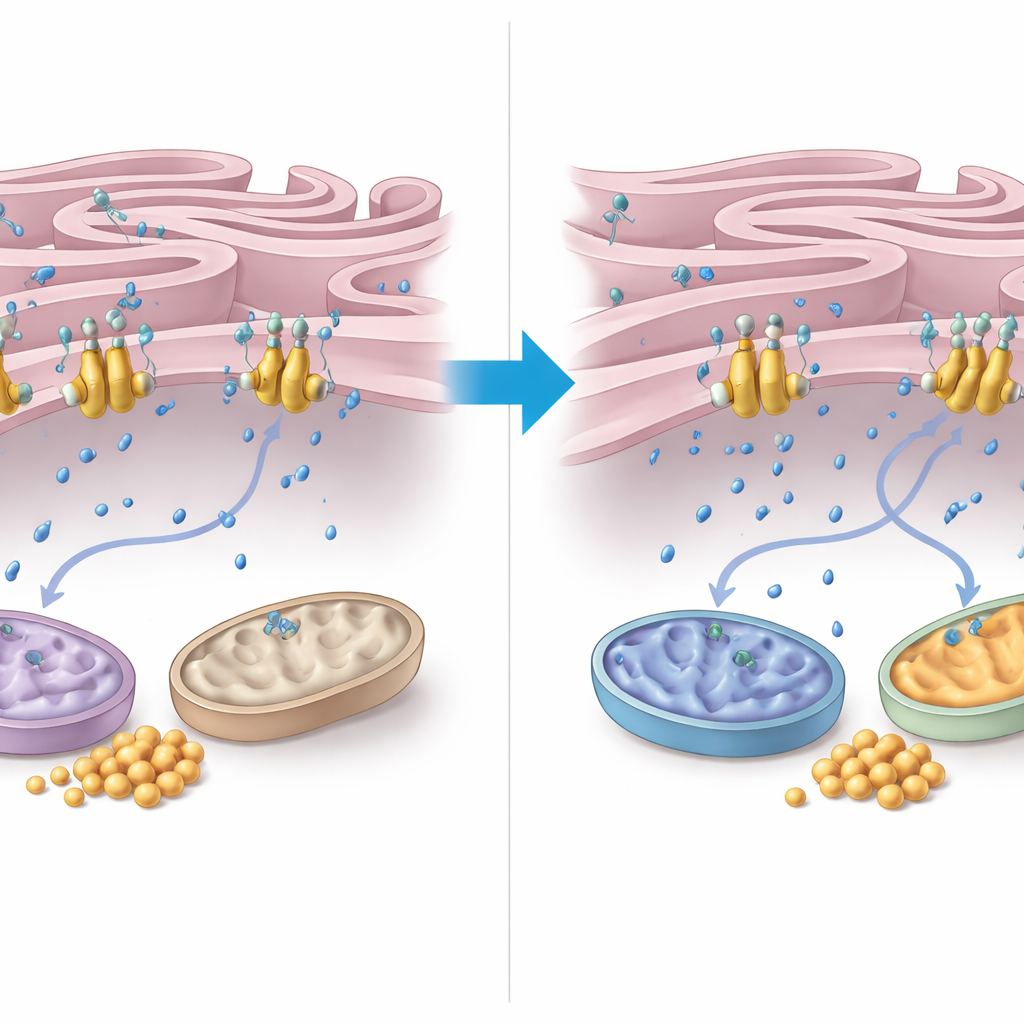
Cómo el calcio transmite la señal de rescate
Para entender cómo ejerce su control FNIP2, los autores se centraron en el manejo del calcio entre el retículo endoplásmico (RE) y las mitocondrias. Descubrieron que FNIP2 se une firmemente a una bomba de calcio en la membrana del RE llamada SERCA2b. En ensayos celulares, eliminar FNIP2 redujo de forma marcada la captación de calcio por el RE, de modo que más calcio permanecía en el citoplasma circundante. Este calcio extra puede estimular enzimas mitocondriales y la respiración. La microscopía electrónica reveló que las células AT presentan mitocondrias deformadas y zonas de contacto entre RE y mitocondrias inusualmente estrechas y extensas —características vinculadas al estrés oxidativo. De forma llamativa, la supresión de FNIP2 no solo normalizó la forma mitocondrial sino que también restauró patrones de contacto RE–mitocondria más típicos, coherentes con una gestión energética más saludable.
Qué significa esto para los pacientes
En conjunto, los hallazgos reinterpretan la ataxia telangiectasia no solo como un trastorno de reparación del ADN sino también como una enfermedad de gestión inadecuada del combustible celular. Sin ATM, la glucosa se utiliza menos para producir energía, las mitocondrias rinden por debajo de lo esperado y el glucógeno obstruye las células en órganos críticos. Al reducir la actividad de FNIP2 —y con ello su estimulación de la bomba de calcio SERCA2b— las células pueden aumentar el calcio citoplasmático lo suficiente para reactivar la actividad mitocondrial, quemar más glucosa y limitar la acumulación de glucógeno. Aunque este trabajo se realizó en células y muestras de tejido, identifica la vía FNIP2–SERCA2b como un objetivo prometedor para terapias dirigidas a mejorar el metabolismo y la supervivencia celular en personas que viven con esta afección compleja.
Cita: Vinciguerra, M., El Kharef, C., Bruhn, C. et al. Targeting the FNIP2-SERCA2b axis improves metabolic and mitochondrial defects in Ataxia Telangiectasia. Cell Death Dis 17, 290 (2026). https://doi.org/10.1038/s41419-026-08507-5
Palabras clave: ataxia telangiectasia, metabolismo celular, mitocondrias, señalización de calcio, acumulación de glucógeno