Clear Sky Science · ru
Целевая модуляция оси FNIP2–SERCA2b улучшает метаболические и митохондриальные нарушения при атаксии-телеангиэктазии
Когда хранитель ДНК клетки также управляет энергией
Атаксия-телеангиэктазия наиболее известна как редкое наследственное заболевание, приводящее к проблемам движения, ослаблению иммунитета и повышенному риску рака. В этом исследовании показано, что помимо защиты ДНК отсутствующий у пациентов белок ATM выполняет роль главного регулятора использования клетками сахаров в качестве источника энергии. При потере ATM клетки сталкиваются не только с повреждением ДНК — они также неправильно перерабатывают глюкозу: накапливают избыток гликогена и лишают митохондрии топлива. Работа указывает на новый молекулярный переключатель, который можно «переключить», чтобы частично восстановить этот энергетический баланс.
Энергетические проблемы при редком заболевании
Исследователи начали с кожных клеток, взятых у людей с атаксией-телеангиэктазией и у здоровых добровольцев. При помощи широкого профилирования метаболитов и измерений потребления кислорода они обнаружили, что клетки AT находятся в состоянии хронического окислительного стресса и плохо сжигают топливо. Ключевые этапы расщепления сахаров (гликолиз) и последующий цикл генерации энергии в митохондриях оба замедлены. В результате клетки AT активнее разрушают белки и нуклеиновые кислоты, как будто добывают альтернативное топливо. Эти дефекты не сводились к дефициту какого‑то одного известного фермента, что указывает на более широкую системную метаболическую неисправность, вызванную потерей ATM.
Когда сахар превращается в тупиковое хранилище
Трассировка меченой глюкозы в живых клетках показала, что меньшая часть поступающего сахара проходила по основным энергетическим путям в клетках AT. Вместо этого накапливались продукты распада, связанные с метаболизмом гликогена. Микроскопия подтвердила, что гликоген — разветвлённая форма запасаемой глюкозы — интенсивно накапливается в фибробластах AT, в индуцированных плюрипотентных стволовых клетках, полученных из этих фибробластов, и в тканях пациентов AT, включая сердце и мозжечок. Нейроны мозжечка, важные для контроля движений, демонстрировали аномальные отложения гликогена и повышенные уровни фермента, синтезирующего гликоген. Этот профиль говорит о том, что при неэффективной работе митохондрий клетки перераспределяют лишнюю глюкозу в запасы, что может дополнительно нагружать уязвимые органы, такие как мозг и сердце.
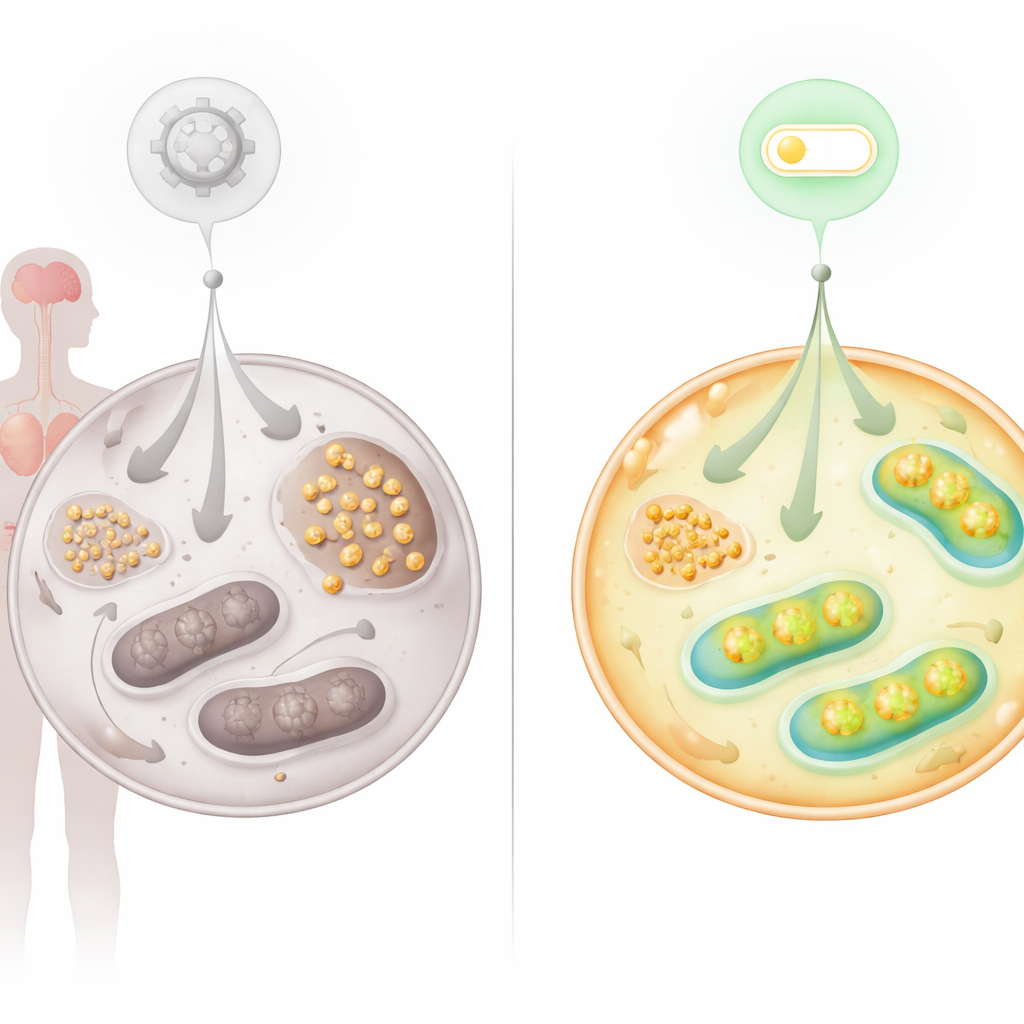
Найден новый переключатель: ось FNIP2–SERCA2b
Поскольку метаболические нарушения явно указывали на сбои в работе митохондрий, команда исследовала белки, известные как регуляторы митохондриальной активности. Они сосредоточились на FNIP2, родственнике FNIP1, который помогает клеткам отвечать на нутритивный и редокс‑стресс. Подавление FNIP2 в клетках AT резко снизило накопление гликогена и восстановило их способность формировать колонии со временем, задержав преждевременное старение, характерное для этих клеток. Подробные биоэнергетические тесты показали, что блокада FNIP2 в клетках AT усиливала гликолиз, повышала митохондриальное дыхание и продукцию АТФ, приближая использование глюкозы к норме, при этом не вызывая чрезмерной компенсации в здоровых клетках.
Как кальций передаёт спасительный сигнал
Чтобы понять, как FNIP2 реализует своё действие, авторы подробно изучили обращение кальция между эндоплазматическим ретикулумом (ЭР) и митохондриями. Они обнаружили, что FNIP2 прочно связывается с кальциевым насосом в мембране ЭР, называемым SERCA2b. В клеточных тестах удаление FNIP2 резко уменьшало поглощение кальция ЭР, что означало увеличение концентрации кальция в окружающем цитоплазматическом пространстве. Этот дополнительный кальций может стимулировать митохондриальные ферменты и дыхание. Сканирующая электронная микроскопия выявила, что в клетках AT митохондрии искажены, а контакты между ЭР и митохондриями необычно плотные и обширные — признаки, связанные с окислительным стрессом. Поразительно, что подавление FNIP2 не только нормализовало форму митохондрий, но и восстанавливало более типичную структуру контактов ЭР–митохондрий, что согласуется с улучшением управления энергией.
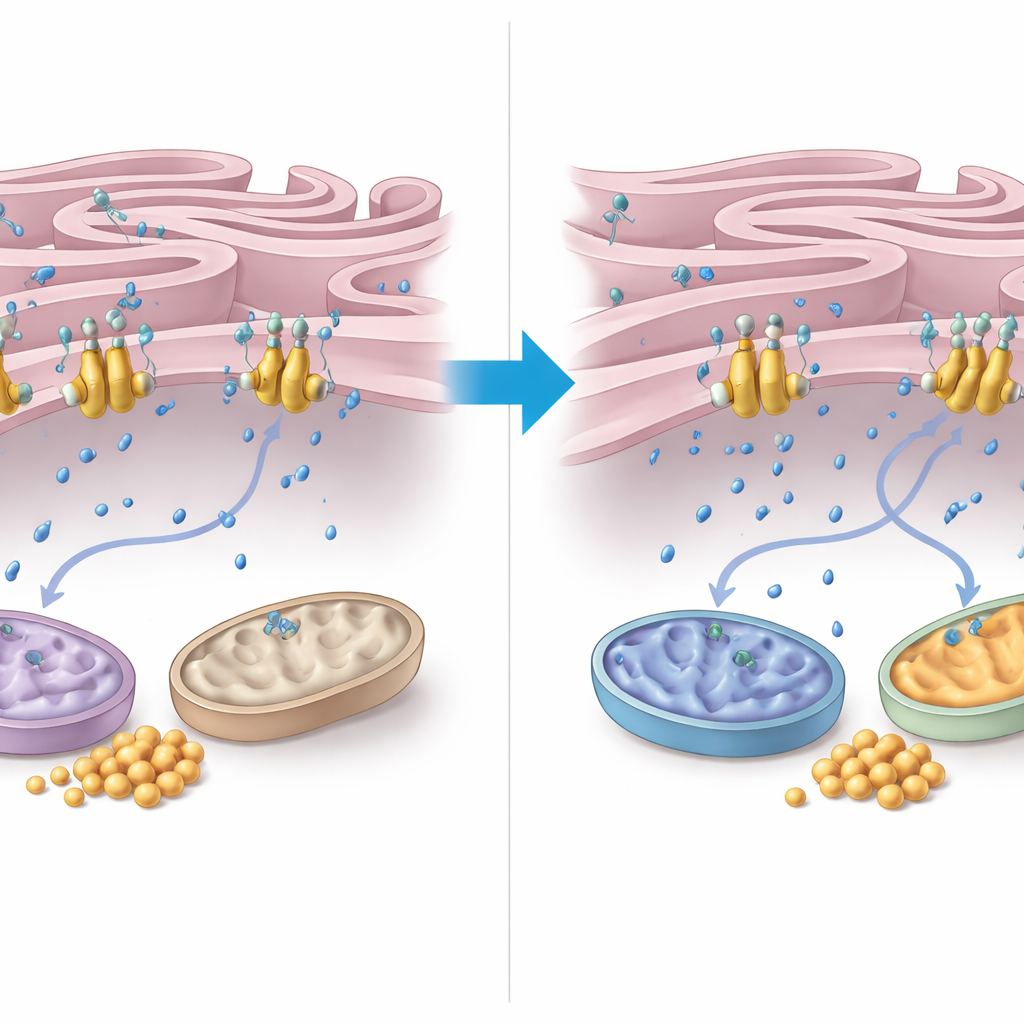
Что это значит для пациентов
В совокупности результаты переводят атаксию‑телеангиэктазию из разряда не только расстройств репарации ДНК, но и заболеваний, связанных с нарушением управления клеточным топливом. При отсутствии ATM глюкоза менее эффективно используется для получения энергии, митохондрии работают слабее, а гликоген закупоривает клетки в критически важных органах. Понижая активность FNIP2 — а значит, ослабляя его стимуляцию кальциевого насоса SERCA2b — можно повысить уровень цитоплазматического кальция настолько, чтобы оживить митохондриальную активность, увеличить сжигание глюкозы и ограничить накопление гликогена. Хотя эти результаты получены в клеточных и тканевых образцах, они выделяют ось FNIP2–SERCA2b как перспективную мишень для терапий, направленных на улучшение метаболизма и выживаемости клеток у людей с этим сложным заболеванием.
Цитирование: Vinciguerra, M., El Kharef, C., Bruhn, C. et al. Targeting the FNIP2-SERCA2b axis improves metabolic and mitochondrial defects in Ataxia Telangiectasia. Cell Death Dis 17, 290 (2026). https://doi.org/10.1038/s41419-026-08507-5
Ключевые слова: атаксия-телеангиэктазия, клеточный метаболизм, митохондрии, кальциевые сигналы, накопление гликогена