Clear Sky Science · de
Die Zielsteuerung der FNIP2‑SERCA2b‑Achse verbessert metabolische und mitochondriale Defekte bei Ataxia telangiectasia
Wenn der DNA‑Wächter der Zelle auch die Energie steuert
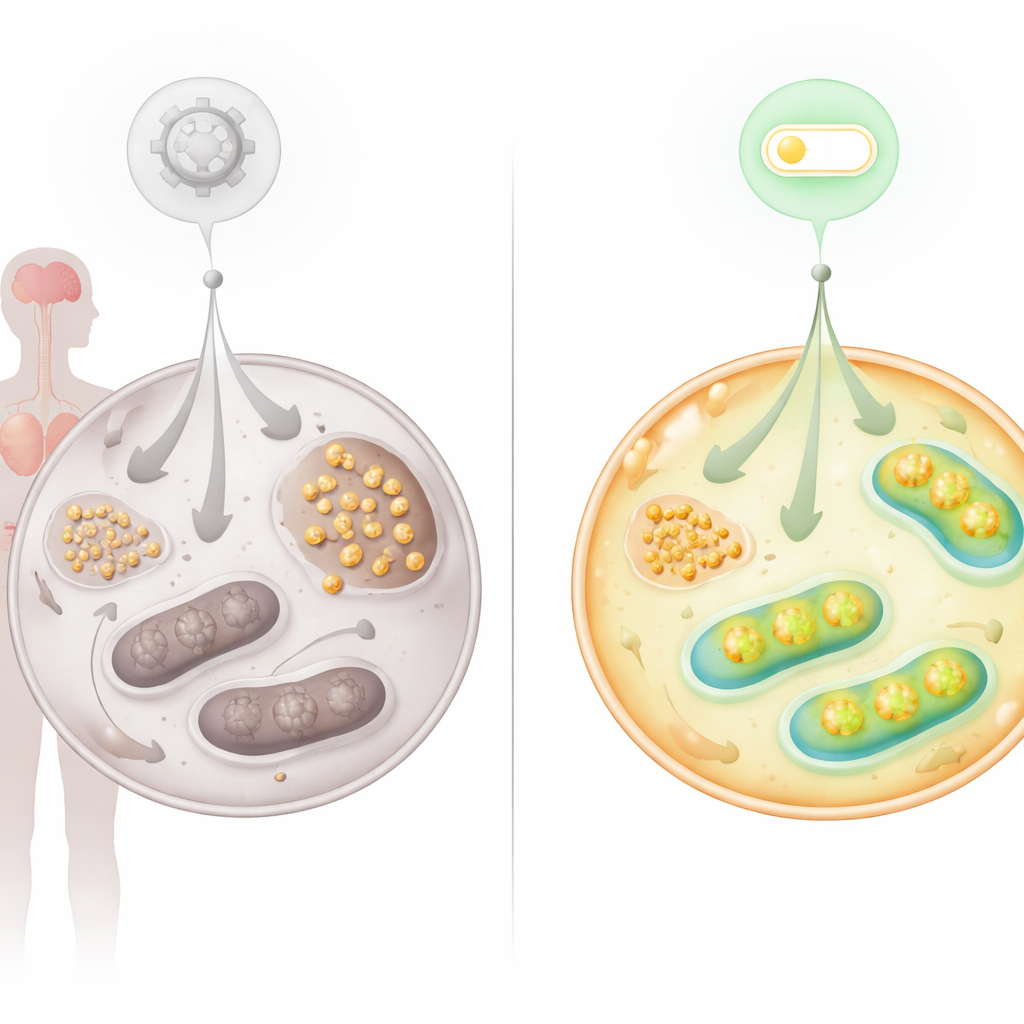
Ataxia telangiectasia ist vor allem als seltene Erbkrankheit bekannt, die Bewegungsstörungen, Immunschwäche und ein erhöhtes Krebsrisiko verursacht. Diese Studie zeigt, dass das fehlende ATM‑Protein bei Betroffenen neben seiner Rolle im DNA‑Schutz auch als zentraler Regulator der zellulären Zucker‑Nutzung fungiert. Geht ATM verloren, haben Zellen nicht nur Probleme mit DNA‑Schäden — sie verarbeiten Glukose falsch, lagern überschüssiges Glykogen an und machen ihre Kraftwerke, die Mitochondrien, regelrecht hungrig. Die Arbeit weist auf einen neuen molekularen Schalter hin, dessen Umlegen das energetische Gleichgewicht teilweise wiederherstellen kann.
Energieprobleme bei einer seltenen Erkrankung
Die Forschenden begannen mit Hautzellen von Menschen mit Ataxia telangiectasia und von gesunden Probanden. Mithilfe umfassender Metabolitprofile und Messungen des Sauerstoffverbrauchs stellten sie fest, dass AT‑Zellen in einem Zustand chronischen oxidativen Stresses leben und Treibstoff schlecht verbrennen. Zentrale Schritte des Zuckerabbaus (Glykolyse) und der nachgeschaltete Energiezyklus in den Mitochondrien liefen beide verlangsamter ab. Infolgedessen bauten AT‑Zellen mehr Proteine und Nukleinsäuren ab als normal, als würden sie alternative Brennstoffe verwerten. Diese Defekte ließen sich nicht einfach durch einen einzelnen bekannten Enzymmangel erklären, was darauf hindeutet, dass der Verlust von ATM ein breiteres, systemisches metabolisches Versagen verursacht.
Wenn Zucker in eine Sackgasse gespeichert wird
Die Verfolgung markierter Glukose in lebenden Zellen zeigte, dass in AT‑Zellen weniger des aufgenommenen Zuckers durch die Hauptenergiepfade floss. Stattdessen häuften sich Abbauprodukte an, die mit dem Glykogenstoffwechsel verbunden sind. Mikroskopische Untersuchungen bestätigten, dass Glykogen — eine verzweigte Speicherform von Glukose — stark in AT‑Fibroblasten, in aus diesen Fibroblasten reprogrammierten Stammzellen und in Geweben von AT‑Patienten, einschließlich Herz und Kleinhirn, akkumuliert. Nervenzellen im Kleinhirn, die für die Bewegungssteuerung wichtig sind, zeigten abnorme Glykogenvorkommen und erhöhte Mengen des Glykogenherstellenden Enzyms. Dieses Muster legt nahe, dass Zellen, wenn Mitochondrien Treibstoff nicht effizient verarbeiten können, überschüssige Glukose in Speicherung umlenken — was wiederum anfällige Organe wie Gehirn und Herz belastet.
Einen neuen Schalter finden: die FNIP2–SERCA2b‑Achse
Da die Stoffwechselprobleme stark auf fehlerhafte Mitochondrien hinwiesen, untersuchte das Team Proteine, die die mitochondriale Aktivität hoch- oder herunterregeln. Sie konzentrierten sich auf FNIP2, einen Verwandten von FNIP1, der Zellen bei Nährstoff‑ und Redoxstress unterstützt. Das Stilllegen von FNIP2 in AT‑Zellen reduzierte die Glykogenansammlung dramatisch und stellte ihre Fähigkeit zur Koloniebildung über Zeit wieder her, wodurch die vorzeitige Seneszenz, die diese Zellen normalerweise prägt, verzögert wurde. Detaillierte bioenergetische Tests zeigten, dass die Hemmung von FNIP2 in AT‑Zellen die Glykolyse verbesserte und die mitochondriale Atmung sowie die ATP‑Produktion erhöhte, sodass die Glukosenutzung näher an den Normalzustand heranrückte, ohne gesunde Zellen zu überkorrigieren.
Wie Kalzium das Rettungssignal überträgt
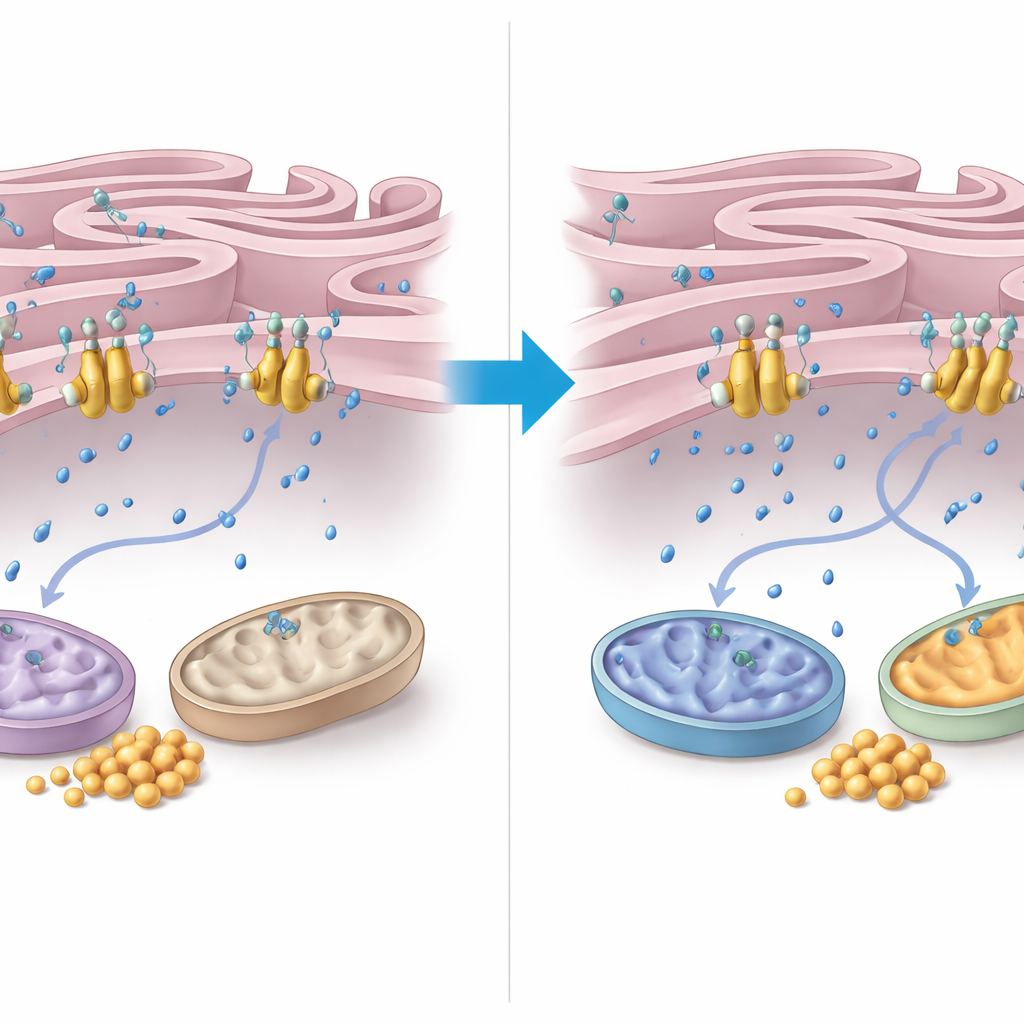
Um zu verstehen, wie FNIP2 diese Kontrolle ausübt, zoomten die Autorinnen und Autoren auf die Kalzium‑Handhabung zwischen dem endoplasmatischen Retikulum (ER) und den Mitochondrien. Sie entdeckten, dass FNIP2 eng an eine Kalziumpumpe in der ER‑Membran bindet, genannt SERCA2b. In zellbasierten Assays verringerte das Entfernen von FNIP2 deutlich die Aufnahme von Kalzium ins ER, sodass mehr Kalzium im umgebenden Zytoplasma verbleibt. Dieses zusätzliche Kalzium kann mitochondriale Enzyme und die Atmung stimulieren. Elektronenmikroskopie zeigte, dass AT‑Zellen verzerrte Mitochondrien und ungewöhnlich enge, ausgedehnte Kontaktzonen zwischen ER und Mitochondrien aufweisen — Merkmale, die mit oxidativem Stress verknüpft sind. Auffällig normalisierte die Unterdrückung von FNIP2 nicht nur die Mitochondrienform, sondern stellte auch typischere ER–Mitochondrien‑Kontaktmuster wieder her, konsistent mit einer gesünderen Energiebewirtschaftung.
Was das für Patientinnen und Patienten bedeutet
In der Summe stellen die Befunde Ataxia telangiectasia nicht nur als Störung der DNA‑Reparatur dar, sondern auch als Krankheit der fehlgesteuerten zellulären Treibstoffwirtschaft. Ohne ATM wird Glukose nur unzureichend zur Energiegewinnung genutzt, Mitochondrien arbeiten schlechter, und Glykogen verstopft Zellen in lebenswichtigen Organen. Durch das Herunterregulieren von FNIP2 — und damit das Abmildern seiner Stimulation der SERCA2b‑Kalziumpumpe — können Zellen den zytoplasmatischen Kalziumspiegel gerade genug anheben, um die mitochondriale Aktivität wiederzubeleben, mehr Glukose zu verbrennen und die Glykogenansammlung zu begrenzen. Obwohl diese Arbeit an Zellen und Gewebeproben durchgeführt wurde, identifiziert sie die FNIP2–SERCA2b‑Achse als vielversprechendes Ziel für Therapien, die darauf abzielen, Stoffwechsel und Zellüberleben bei Menschen mit dieser komplexen Erkrankung zu verbessern.
Zitation: Vinciguerra, M., El Kharef, C., Bruhn, C. et al. Targeting the FNIP2-SERCA2b axis improves metabolic and mitochondrial defects in Ataxia Telangiectasia. Cell Death Dis 17, 290 (2026). https://doi.org/10.1038/s41419-026-08507-5
Schlüsselwörter: ataxia telangiectasia, zellulärer Stoffwechsel, Mitochondrien, Kalziumsignalgebung, Glykogenansammlung