Clear Sky Science · pt
Alvo no eixo FNIP2-SERCA2b melhora defeitos metabólicos e mitocondriais na Ataxia Telangiectasia
Quando o guardião do DNA da célula também gerencia a energia
A ataxia telangiectasia é mais conhecida como uma doença genética rara que causa problemas de movimento, fragilidade do sistema imunológico e risco de câncer. Este estudo mostra que, além de proteger nosso DNA, a proteína ATM ausente nesses pacientes também atua como um gerente central de como as células usam a glicose para gerar energia. Quando o ATM se perde, as células não só têm dificuldade com danos ao DNA — elas também manejam mal a glicose, acumulando glicogênio em excesso e privando suas usinas de energia, as mitocôndrias. O trabalho aponta para um novo interruptor molecular que pode ser acionado para restaurar parcialmente esse equilíbrio energético.
Problemas de energia em uma doença rara
Os pesquisadores começaram com células da pele retiradas de pessoas com ataxia telangiectasia e de voluntários saudáveis. Usando um amplo perfil de metabólitos e medições de consumo de oxigênio, eles descobriram que as células AT vivem em um estado de estresse oxidativo crônico e queimam combustível de forma ineficiente. Etapas-chave da quebra da glicose (glicólise) e o ciclo gerador de energia nas mitocôndrias estavam lentos. Como resultado, as células AT degradaram proteínas e ácidos nucleicos mais do que o normal, como se estivessem catando combustíveis alternativos. Esses defeitos não se explicavam simplesmente por uma deficiência de uma única enzima conhecida, indicando que a perda de ATM causa uma falha metabólica mais ampla, em nível de sistema.
Quando a glicose vira um depósito sem saída
Rastrear glicose marcada em células vivas revelou que menos da glicose que entrava seguia pelos principais caminhos energéticos nas células AT. Em vez disso, produtos de degradação relacionados ao metabolismo do glicogênio se acumularam. A microscopia confirmou que o glicogênio — uma forma ramificada de armazenamento de glicose — se acumulou intensamente em fibroblastos AT, em células-tronco reprogramadas a partir desses fibroblastos e em tecidos de pacientes AT, incluindo coração e cerebelo. Células nervosas do cerebelo, cruciais para o controle do movimento, mostraram depósitos anormais de glicogênio e níveis elevados da enzima que sintetiza glicogênio. Esse padrão sugere que, quando as mitocôndrias não conseguem processar o combustível com eficiência, as células desviam o excesso de glicose para armazenamento, o que pode, por sua vez, sobrecarregar órgãos vulneráveis como cérebro e coração.
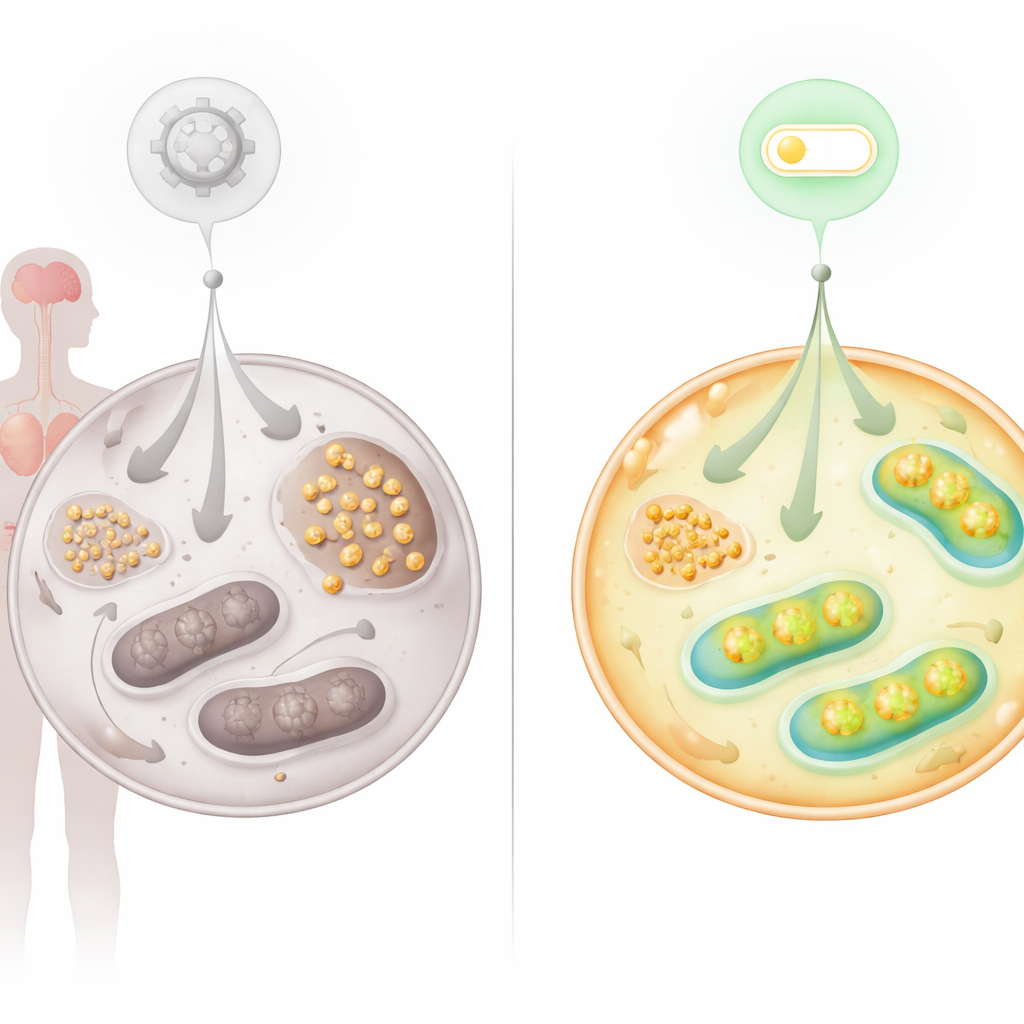
Encontrando um novo interruptor: o eixo FNIP2–SERCA2b
Como os problemas metabólicos apontavam fortemente para mitocôndrias defeituosas, a equipe investigou proteínas conhecidas por ajustar a atividade mitocondrial para cima ou para baixo. Eles se concentraram em FNIP2, um parente de FNIP1, que ajuda as células a responder ao estresse nutricional e redox. Silenciar FNIP2 em células AT reduziu dramaticamente o acúmulo de glicogênio e restaurou a capacidade dessas células de formar colônias ao longo do tempo, retardando a senescência prematura que normalmente afeta essas células. Testes bioenergéticos detalhados mostraram que bloquear FNIP2 em células AT aumentou a glicólise e elevou a respiração mitocondrial e a produção de ATP, aproximando o uso de glicose do normal sem causar uma correção excessiva em células saudáveis.
Como o cálcio transmite o sinal de resgate
Para entender como o FNIP2 exerce esse controle, os autores aprofundaram-se no manejo do cálcio entre o retículo endoplasmático (RE) e as mitocôndrias. Eles descobriram que FNIP2 se liga fortemente a uma bomba de cálcio na membrana do RE chamada SERCA2b. Em ensaios celulares, remover FNIP2 reduziu acentuadamente a captação de cálcio pelo RE, o que significa que mais cálcio permaneceu no citoplasma circundante. Esse cálcio extra pode estimular enzimas mitocondriais e a respiração. Microscopia eletrônica revelou que células AT têm mitocôndrias distorcidas e zonas de contato entre RE e mitocôndrias incomumente estreitas e extensas — características ligadas ao estresse oxidativo. De forma marcante, a supressão de FNIP2 não apenas normalizou a forma mitocondrial, mas também restaurou padrões mais típicos de contato RE–mitocôndria, coerentes com um gerenciamento de energia mais saudável.
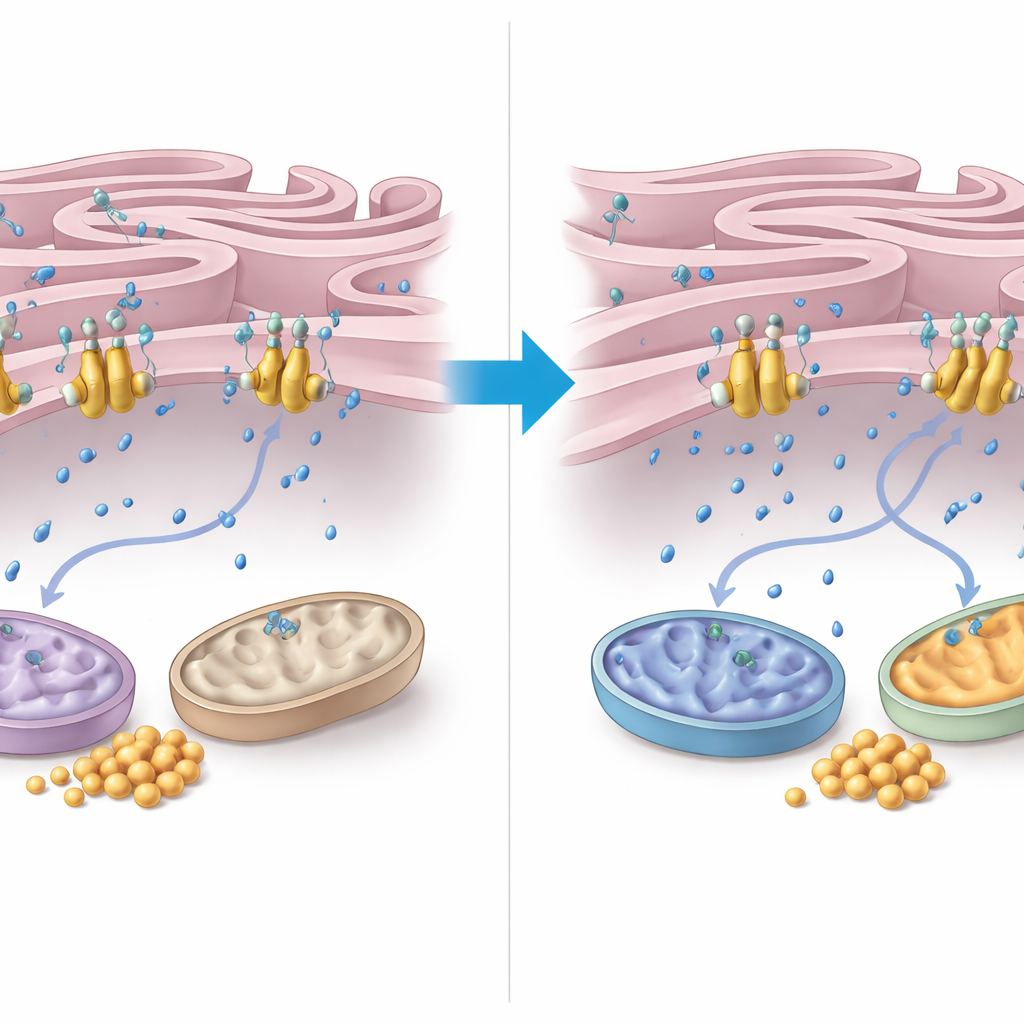
O que isso significa para os pacientes
No conjunto, os achados reinterpretam a ataxia telangiectasia não apenas como um transtorno de reparo de DNA, mas também como uma doença de gerenciamento inadequado de combustível celular. Sem ATM, a glicose é subutilizada para gerar energia, as mitocôndrias têm desempenho reduzido e o glicogênio entope as células em órgãos críticos. Ao reduzir a atividade de FNIP2 — e assim atenuar sua estimulação da bomba de cálcio SERCA2b — as células podem elevar o cálcio citoplasmático na medida certa para reavivar a atividade mitocondrial, queimar mais glicose e limitar o acúmulo de glicogênio. Embora este trabalho tenha sido realizado em células e amostras de tecido, ele identifica a via FNIP2–SERCA2b como um alvo promissor para terapias destinadas a melhorar o metabolismo e a sobrevivência celular em pessoas que vivem com essa condição complexa.
Citação: Vinciguerra, M., El Kharef, C., Bruhn, C. et al. Targeting the FNIP2-SERCA2b axis improves metabolic and mitochondrial defects in Ataxia Telangiectasia. Cell Death Dis 17, 290 (2026). https://doi.org/10.1038/s41419-026-08507-5
Palavras-chave: ataxia telangiectasia, metabolismo celular, mitocôndrias, sinalização de cálcio, acúmulo de glicogênio