Clear Sky Science · pl
Celowanie w oś FNIP2–SERCA2b poprawia defekty metaboliczne i mitochondrialne w ataksji teleangiektazji
Gdy strażnik DNA komórki zarządza też energetyką
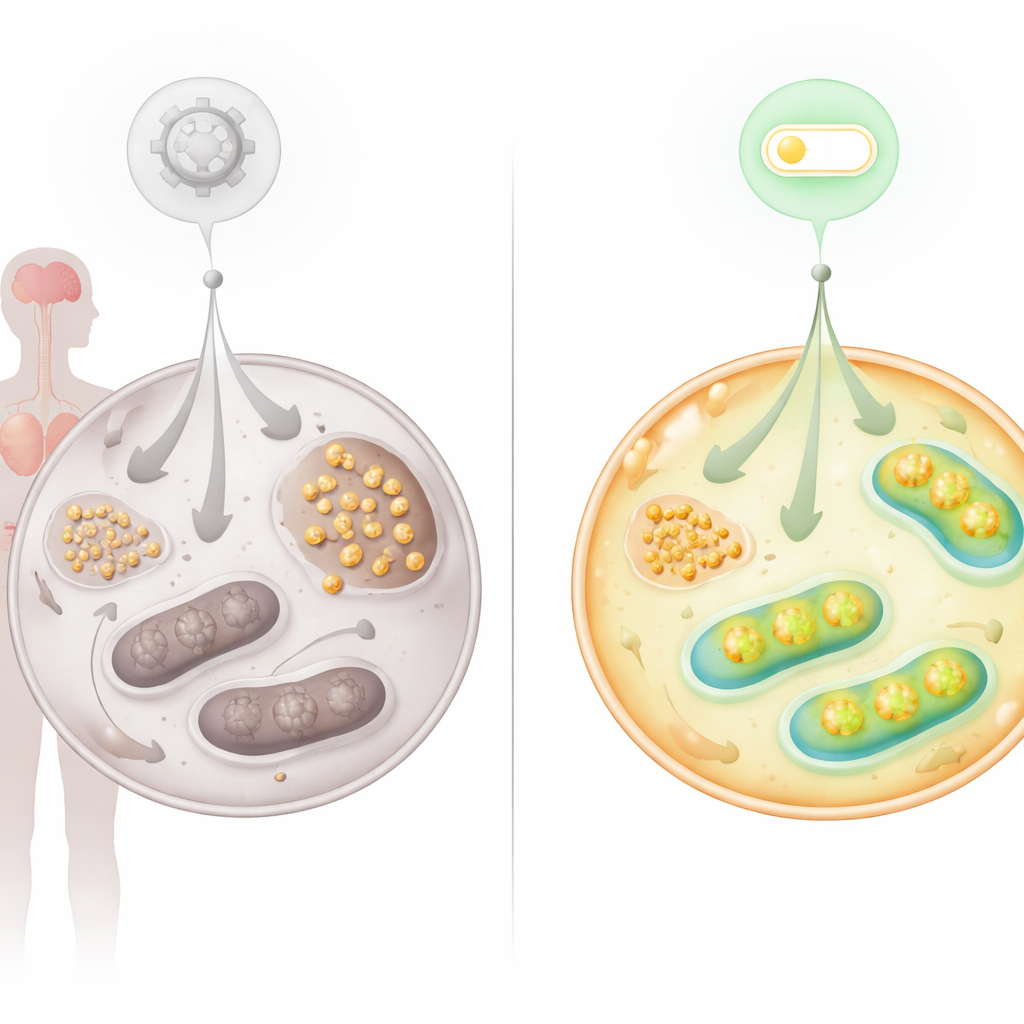
Ataksja teleangiektazja jest najbardziej znana jako rzadka choroba genetyczna powodująca problemy z ruchem, osłabienie odporności i zwiększone ryzyko nowotworów. Badanie pokazuje, że oprócz ochrony materiału genetycznego, brak białka ATM u tych pacjentów pełni także rolę głównego menedżera wykorzystania cukru przez komórki do produkcji energii. Gdy ATM jest nieobecny, komórki nie tylko mają trudności z naprawą DNA — źle gospodarują glukozą, odkładając nadmiar w postaci glikogenu i pozbawiając mitochondria paliwa. Praca wskazuje nowy przełącznik molekularny, którego manipulacja może częściowo przywrócić równowagę energetyczną.
Problemy z energią w rzadkiej chorobie
Naukowcy rozpoczęli od komórek skóry pobranych od osób z ataksją teleangiektazją i od zdrowych ochotników. Przy użyciu szerokiego profilowania metabolitów i pomiarów zużycia tlenu odkryli, że komórki AT żyją w stanie przewlekłego stresu oksydacyjnego i słabo spalają paliwo. Kluczowe etapy rozkładu cukru (glikoliza) oraz cykl energetyczny w mitochondriach były spowolnione. W efekcie komórki AT bardziej niż zwykle rozkładały białka i kwasy nukleinowe, jakby pozyskiwały alternatywne źródła energii. Te defekty nie były prostym odtworzeniem jednego znanego niedoboru enzymatycznego, co wskazuje, że utrata ATM powoduje szerszą, systemową niewydolność metaboliczną.
Gdy cukier trafia w ślepy zaułek magazynu
Śledzenie znakowanej glukozy w żywych komórkach ujawniło, że mniej przychodzącego cukru przepływa przez główne szlaki energetyczne w komórkach AT. Zamiast tego kumulowały się produkty rozkładu związane z metabolizmem glikogenu. Mikroskopia potwierdziła, że glikogen — rozgałęziona forma zapasowa glukozy — gromadzi się intensywnie w fibroblastach AT, w komórkach macierzystych przeprogramowanych z tych fibroblastów oraz w tkankach pacjentów z AT, w tym w sercu i móżdżku. Komórki nerwowe móżdżku, istotne dla kontroli ruchu, wykazywały nieprawidłowe odkładanie glikogenu i podwyższone poziomy enzymu syntetyzującego glikogen. Ten obraz sugeruje, że gdy mitochondria nie potrafią efektywnie przetwarzać paliwa, komórki kierują nadmiar glukozy do magazynowania, co z kolei może obciążać wrażliwe narządy, takie jak mózg i serce.
Odnalezienie nowego przełącznika: oś FNIP2–SERCA2b
Ponieważ problemy metaboliczne jednoznacznie wskazywały na dysfunkcję mitochondriów, zespół przyjrzał się białkom znanym z regulacji aktywności mitochondrialnej. Skoncentrowano się na FNIP2, krewniaku FNIP1, który pomaga komórkom reagować na stres związany z odżywianiem i zmianami redoks. Wyciszenie FNIP2 w komórkach AT drastycznie zmniejszyło nagromadzenie glikogenu i przywróciło ich zdolność do tworzenia kolonii w czasie, opóźniając przedwczesne starzenie się, które zwykle dotyka te komórki. Szczegółowe testy bioenergetyczne wykazały, że blokada FNIP2 w komórkach AT poprawiła glikolizę oraz zwiększyła oddychanie mitochondrialne i produkcję ATP, przybliżając wykorzystanie glukozy do normy, bez nadmiernej korekty w komórkach zdrowych.
Jak wapń przekazuje sygnał ratunkowy
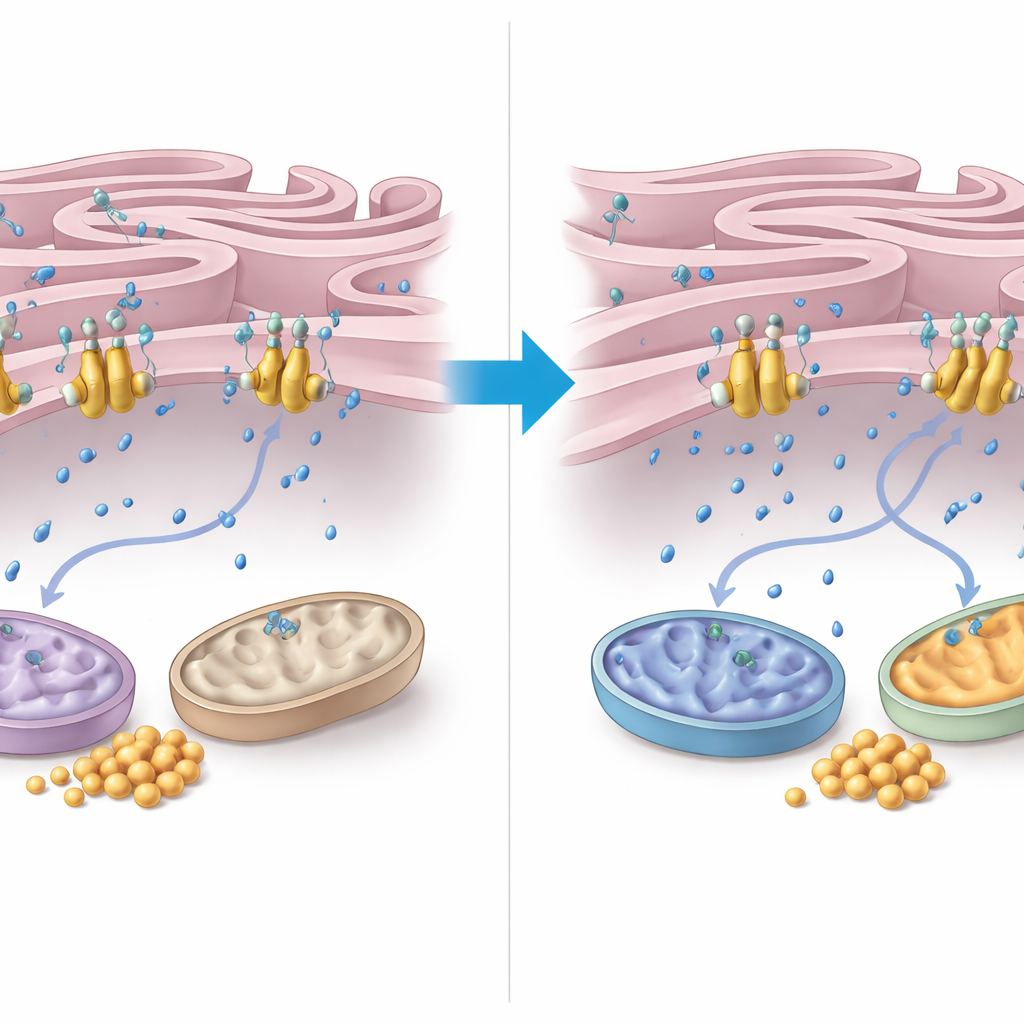
Aby zrozumieć, jak FNIP2 wywiera tę kontrolę, autorzy przyjrzeli się gospodarcze wapnia między siateczką śródplazmatyczną (ER) a mitochondriami. Odkryli, że FNIP2 ściśle wiąże się z pompą wapniową w błonie ER o nazwie SERCA2b. W testach komórkowych usunięcie FNIP2 wyraźnie zmniejszyło wychwyt wapnia przez ER, co oznacza, że więcej wapnia pozostawało w otaczającej cytoplazmie. Ten dodatkowy wapń może pobudzać enzymy mitochondrialne i oddychanie. Mikroskopia elektronowa ujawniła, że komórki AT mają zniekształcone mitochondria i niezwykle ciasne, rozległe strefy kontaktowe między ER a mitochondriami — cechy związane ze stresem oksydacyjnym. Co uderzające, tłumienie FNIP2 nie tylko normalizowało kształt mitochondriów, lecz także przywracało bardziej typowy wzorzec kontaktów ER–mitochondria, co koresponduje ze zdrowszym zarządzaniem energią.
Co to oznacza dla pacjentów
Podsumowując, wyniki przekształcają obraz ataksji teleangiektazji z zaburzenia naprawy DNA w chorobę niewłaściwego zarządzania paliwem komórkowym. Bez ATM glukoza jest niedostatecznie wykorzystywana do produkcji energii, mitochondria działają poniżej możliwości, a glikogen zatyka komórki w kluczowych narządach. poprzez osłabienie FNIP2 — a w konsekwencji jego stymulacji pompy wapniowej SERCA2b — komórki mogą podnieść stężenie wapnia w cytoplazmie na tyle, by ożywić aktywność mitochondrialną, spalać więcej glukozy i ograniczać odkładanie glikogenu. Chociaż prace te przeprowadzono na komórkach i próbkach tkankowych, identyfikują one ścieżkę FNIP2–SERCA2b jako obiecujący cel terapii mających na celu poprawę metabolizmu i przeżywalności komórek u osób żyjących z tą złożoną chorobą.
Cytowanie: Vinciguerra, M., El Kharef, C., Bruhn, C. et al. Targeting the FNIP2-SERCA2b axis improves metabolic and mitochondrial defects in Ataxia Telangiectasia. Cell Death Dis 17, 290 (2026). https://doi.org/10.1038/s41419-026-08507-5
Słowa kluczowe: ataksja teleangiektazja, metabolizm komórkowy, mitochondria, sygnalizacja wapniowa, nagromadzenie glikogenu