Clear Sky Science · it
Mirare l’asse FNIP2-SERCA2b migliora i difetti metabolici e mitocondriali nell’atassia-telangiectasia
Quando il guardiano del DNA della cellula gestisce anche l’energia
L’atassia-telangiectasia è nota soprattutto come una rara malattia genetica che provoca problemi di movimento, deficit immunitario e aumentato rischio di cancro. Questo studio mostra che, oltre a proteggere il nostro DNA, la proteina ATM assente in questi pazienti agisce anche come un regolatore principale dell’uso del glucosio per l’energia. In assenza di ATM, le cellule non solo faticano a riparare il DNA: gestiscono male il glucosio, accumulando glicogeno in eccesso e privando di carburante i loro generatori di energia, i mitocondri. Il lavoro individua un nuovo interruttore molecolare che può essere azionato per ripristinare parzialmente questo equilibrio energetico.
Problemi energetici in una malattia rara
I ricercatori hanno iniziato con cellule della pelle prelevate da persone con atassia-telangiectasia e da volontari sani. Utilizzando un ampio profilo di metaboliti e misure del consumo di ossigeno, hanno scoperto che le cellule AT vivono in uno stato cronico di stress ossidativo e bruciano il combustibile in modo inefficiente. Passaggi chiave della degradazione del glucosio (glicolisi) e il successivo ciclo di produzione di energia nei mitocondri erano entrambi rallentati. Di conseguenza, le cellule AT degradavano proteine e acidi nucleici più del normale, come se recuperassero combustibili alternativi. Questi difetti non erano semplicemente la ripetizione di una singola nota carenza enzimatica, indicando che la perdita di ATM causa un guasto metabolico più ampio a livello di sistema.
Quando lo zucchero diventa uno stoccaggio senza uscita
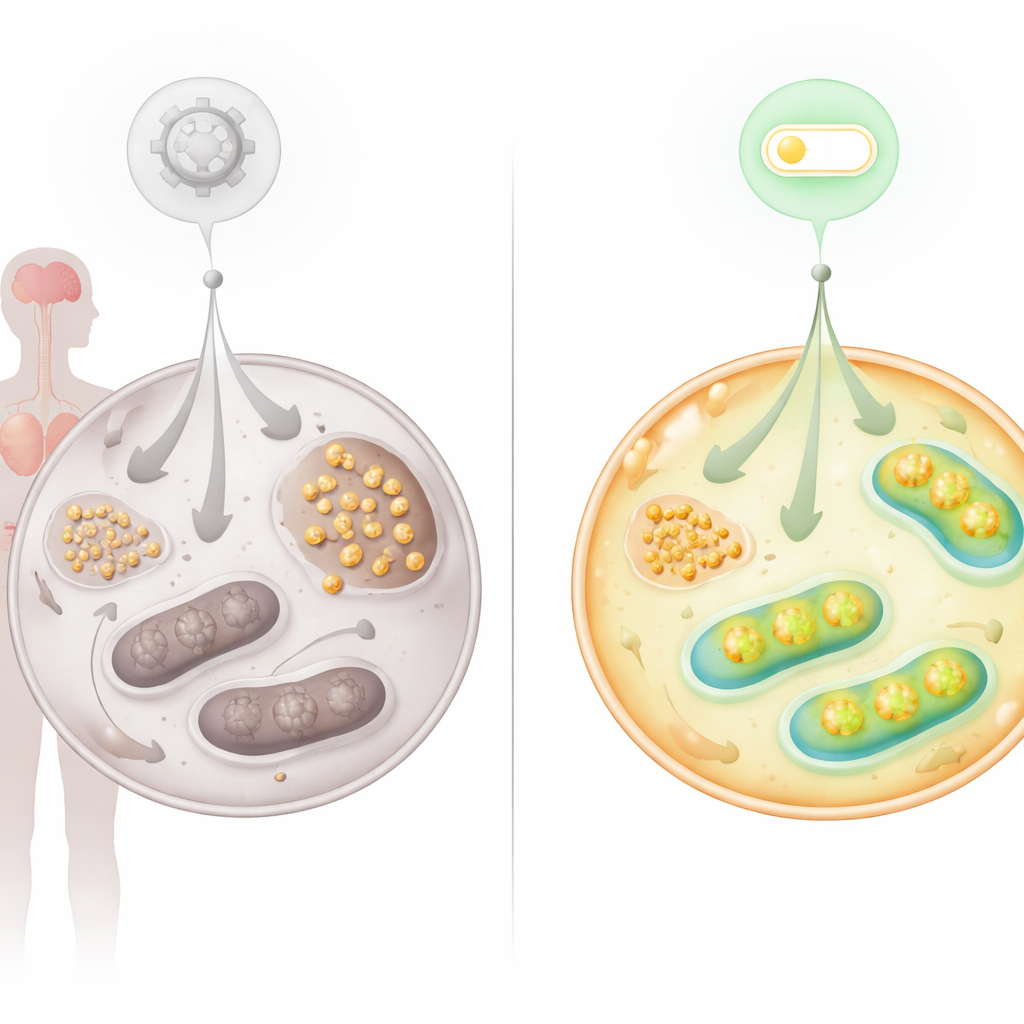
Tracciando il glucosio marcato all’interno di cellule vive è emerso che meno zucchero in ingresso fluiva attraverso le principali vie energetiche nelle cellule AT. Al contrario, si accumulavano prodotti di degradazione legati al metabolismo del glicogeno. La microscopia ha confermato che il glicogeno — una forma ramificata di immagazzinamento del glucosio — si accumulava pesantemente nei fibroblasti AT, nelle cellule staminali riprogrammate da quei fibroblasti e nei tessuti di pazienti AT, incluso cuore e cervelletto. Le cellule nervose del cervelletto, cruciali per il controllo del movimento, mostravano depositi di glicogeno anomali e livelli aumentati dell’enzima che sintetizza il glicogeno. Questo quadro suggerisce che quando i mitocondri non riescono a processare efficacemente il combustibile, le cellule deviano il glucosio in eccesso verso lo stoccaggio, il che può a sua volta mettere sotto stress organi vulnerabili come cervello e cuore.
Individuare un nuovo interruttore: l’asse FNIP2–SERCA2b
Poiché i problemi metabolici indicavano fortemente mitocondri difettosi, il gruppo ha esaminato proteine note per modulare l’attività mitocondriale. Si sono concentrati su FNIP2, cugino di FNIP1, che aiuta le cellule a rispondere allo stress nutrizionale e redox. Silenziare FNIP2 nelle cellule AT ha ridotto drasticamente l’accumulo di glicogeno e ha ripristinato la loro capacità di formare colonie nel tempo, ritardando la senescenza prematura che di solito affligge queste cellule. Test bioenergetici dettagliati hanno mostrato che bloccare FNIP2 nelle cellule AT ha potenziato la glicolisi e aumentato la respirazione mitocondriale e la produzione di ATP, avvicinando l’utilizzo del glucosio a livelli più normali senza ipercompensare nelle cellule sane.
Come il calcio trasmette il segnale di recupero
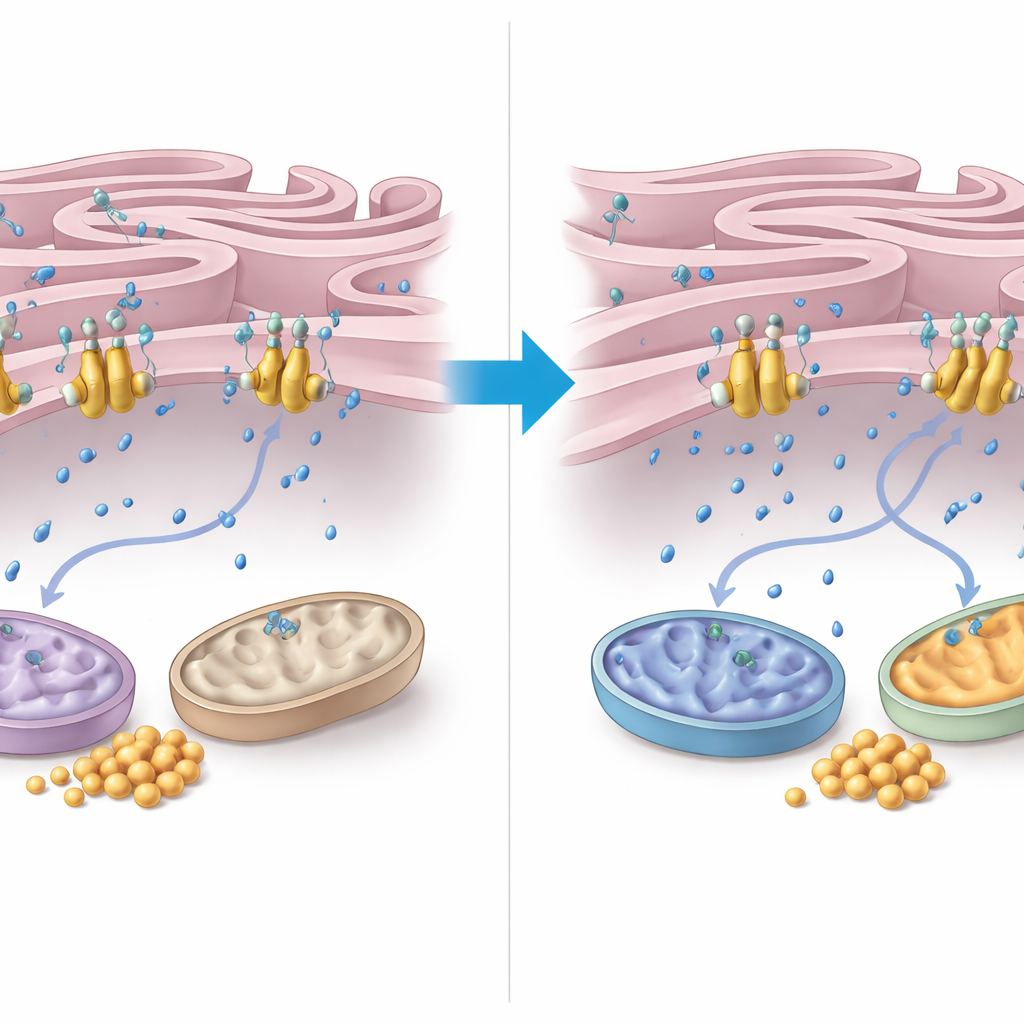
Per capire come FNIP2 eserciti questo controllo, gli autori si sono focalizzati sulla gestione del calcio tra reticolo endoplasmatico (RE) e mitocondri. Hanno scoperto che FNIP2 si lega saldamente a una pompa del calcio nella membrana del RE chiamata SERCA2b. In saggi cellulari, la rimozione di FNIP2 riduceva nettamente l’assorbimento di calcio da parte del RE, lasciando più calcio nel citoplasma circostante. Questo calcio in eccesso può stimolare enzimi mitocondriali e la respirazione. La microscopia elettronica ha rivelato che le cellule AT presentano mitocondri distorti e zone di contatto tra RE e mitocondri insolitamente strette ed estese — caratteristiche associate allo stress ossidativo. Colpisce che la soppressione di FNIP2 non solo normalizzasse la forma mitocondriale ma ripristinasse anche modelli di contatto RE–mitocondri più tipici, coerenti con una gestione energetica più sana.
Che cosa significa per i pazienti
Nel complesso, i risultati reinterpretano l’atassia-telangiectasia non solo come un disturbo della riparazione del DNA ma anche come una malattia di cattiva gestione del carburante cellulare. In assenza di ATM, il glucosio è sottoutilizzato per l’energia, i mitocondri funzionano sotto tono e il glicogeno ostruisce le cellule in organi critici. Riducendo l’attività di FNIP2 — e quindi attenuandone la stimolazione della pompa del calcio SERCA2b — le cellule possono aumentare il calcio citoplasmatico quanto basta per ravvivare l’attività mitocondriale, bruciare più glucosio e limitare l’accumulo di glicogeno. Sebbene questo lavoro sia stato condotto in cellule e campioni di tessuto, identifica la via FNIP2–SERCA2b come un obiettivo promettente per terapie volte a migliorare il metabolismo e la sopravvivenza cellulare nelle persone che convivono con questa condizione complessa.
Citazione: Vinciguerra, M., El Kharef, C., Bruhn, C. et al. Targeting the FNIP2-SERCA2b axis improves metabolic and mitochondrial defects in Ataxia Telangiectasia. Cell Death Dis 17, 290 (2026). https://doi.org/10.1038/s41419-026-08507-5
Parole chiave: atassia telangiectasia, metabolismo cellulare, mitocondri, segnalazione del calcio, accumulo di glicogeno