Clear Sky Science · sv
Kardiomyocyt‑producerat TGFB3 dämpar hjärtfibros och bevarar hjärtfunktionen vid hjärtsvikt
Varför detta spelar roll för personer med svaga hjärtan
Hjärtsvikt är ett vanligt och allvarligt tillstånd där hjärtat gradvis förlorar sin förmåga att pumpa blod. En huvudorsak är ärrbildning i hjärtat, så kallad fibros, som gör hjärtmuskeln stelare och förvärrar symtomen. Denna studie avslöjar en överraskande skyddande signal som hjärtats egna muskelceller producerar och som kan bromsa denna ärrbildning och hjälpa till att bevara hjärtfunktionen — vilket pekar mot en ny, mer precis behandlingsstrategi för hjärtsvikt.
En närmare titt på ärrbildning i hjärtat
Vid hjärtsvikt ”slits” inte hjärtat bara ut. Istället omformas dess vävnad: muskelceller förstoras eller dör, och bindvävsceller avsätter överskott av kollagen som bildar hårda, ärrliknande områden. Under lång tid har en kraftfull familj av signalsubstanser, känd som TGF‑beta, kopplats till denna ärrbildning. Att blockera alla former av TGF‑beta visade sig dock vara ett för grovt grepp och gav allvarliga biverkningar eftersom dessa molekyler också medverkar i normal reparation och immunbalans. Den nya studien undersöker om en särskild familjemedlem, kallad TGF‑beta3, kanske snarare hjälper hjärtat att försvara sig mot okontrollerad fibros än att driva den.
Upptäckt av en skyddande signal i sjuka hjärtan
Forskarna undersökte hjärtvävnad från patienter med avancerad hjärtsvikt och från personer utan hjärtsvikt, samt flera musemodeller som efterliknar trycköverbelastning på hjärtat. De fann att nivåerna av TGF‑beta3 konsekvent var högre i sviktande hjärtan än i friska, både i vävnaden och i blodomloppet. Viktigt är att denna ökning inte främst kom från de vanliga ärrbildande cellerna, utan från kardiomyocyterna — muskelcellerna som faktiskt kontraherar för att pumpa blod. Patienter med högre blodnivåer av TGF‑beta3 tenderade också att ha högre nivåer av en standardmarkör för hjärtsvikt, vilket tyder på att denna molekyl speglar hur intensivt hjärtat belastas.
Släcka signalen i hjärtmuskelceller
För att testa om det kardiomyocyt‑producerade TGF‑beta3 var hjälpsamt eller skadligt konstruerade teamet möss där genen för TGF‑beta3 selektivt kunde tas bort endast i hjärtmuskelceller. Under normala, vilande förhållanden såg dessa möss ut att ha hjärtan som liknade deras syskon både i utseende och funktion. Men när djuren utsattes för en procedur som förträngde huvudartären från hjärtat — ett standardförfarande för att inducera kronisk trycköverbelastning och så småningom hjärtsvikt — blev skillnaderna slående. Möss utan TGF‑beta3 i sina kardiomyocyter utvecklade sämre pumpfunktion, större hjärtan och avsevärt mer kollagenuppbyggnad mellan muskelfibrerna än kontrollmöss som utsattes för samma stress.
Hur hjärtmuskelceller kommunicerar med ärrbildande celler
För att gå till botten med mekanismen jämförde forskarna genaktivitet i hjärtan med och utan kardiomyocyt‑TGF‑beta3. Hjärtan som saknade denna signal visade en kraftig ökning av två väletablerade drivkrafter för fibros, CTGF och SERPINE1, vilka huvudsakligen produceras av fibroblaster — cellerna som ansvarar för att bilda bindväv. I cellkulturer, när fibroblaster exponerades för den klassiska ärrbildande signalen TGF‑beta1, aktiverade de dessa fibrotiska gener och intog ett aktiverat, matrixproducerande tillstånd. Tillsatt TGF‑beta3 dämpade emellertid detta svar. Fibroblaster badade i sekret från TGF‑beta3‑defekta kardiomyocyter blev mer aktiverade än de som exponerades för sekret från normala kardiomyocyter, vilket bekräftar att muskelcellerna normalt utsöndrar en faktor som begränsar fibroblastöveraktivitet.
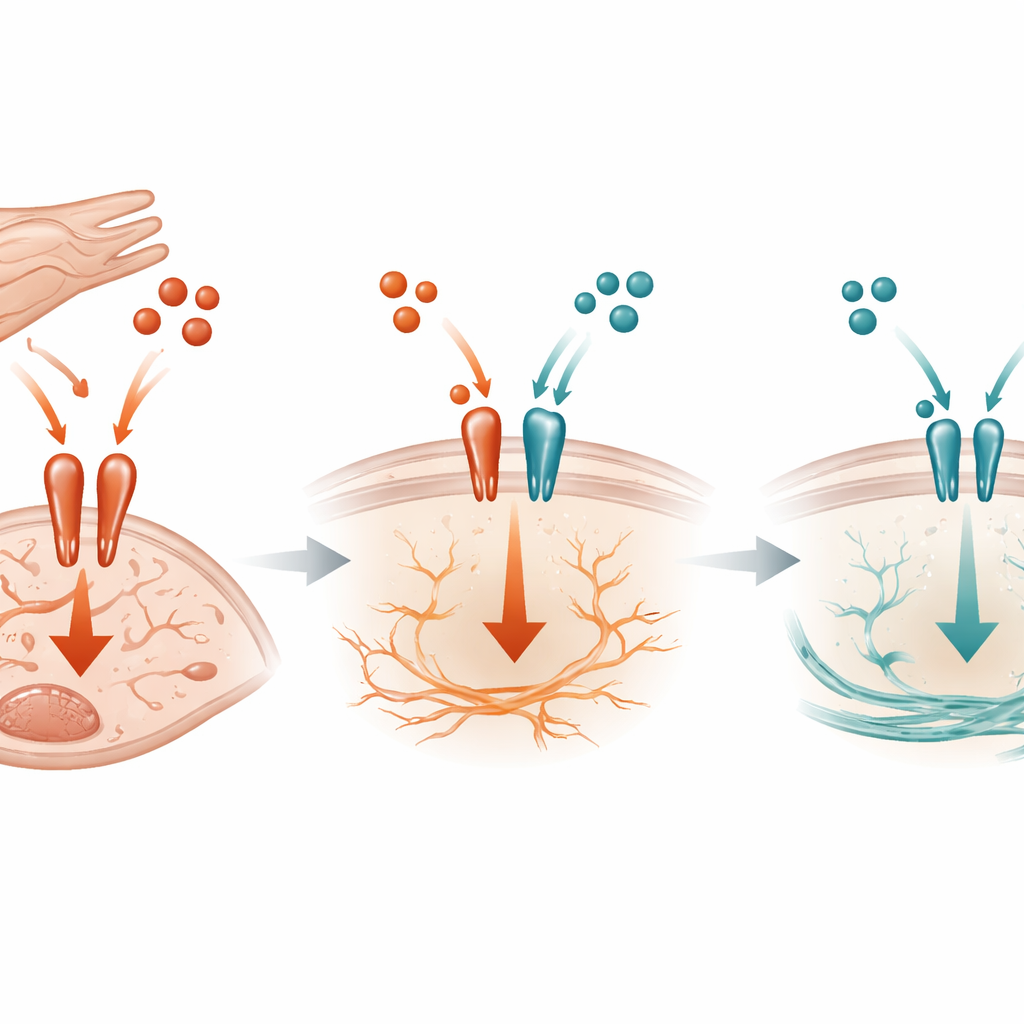
En molekylär dragkamp vid cellytan
På molekylär nivå visar studien att TGF‑beta3 fungerar som en konkurrerande broms mot den mer aggressiva TGF‑beta1‑signalen. Båda molekylerna använder samma receptor komplex på fibroblastytor för att utlösa aktivitet inne i cellen. Biokemiska experiment visade att TGF‑beta3 kan binda till dessa receptorer och begränsa hur mycket TGF‑beta1 kan haka på, vilket minskar aktiveringen av en central brytare inne i cellen kallad SMAD3. När kardiomyocyt‑härlett TGF‑beta3 saknas hos möss under trycköverbelastning ökar SMAD3‑aktiveringen, CTGF och SERPINE1 stiger och fibrosen accelererar. Noterbart är att denna effekt inte berodde på en annan känd inhibitor inom vägen, vilket tyder på att enkel konkurrens om receptorn är en central del av den skyddande mekanismen.
Vad detta kan innebära för framtida behandlingar
För icke‑specialisten är huvudbudskapet att det sviktande hjärtat inte bara är ett offer för ärrbildning; det försöker också skydda sig självt. Hjärtmuskelceller frigör TGF‑beta3 som ett lokalt, inbyggt försvar som drar emot starkare pro‑ärrsignaler och hindrar bindväven från att gå i övervarv. När denna skyddande signal saknas förvärras fibrosen och hjärtfunktionen försämras snabbare. Istället för att blockera hela TGF‑beta‑systemet kan framtida terapier sikta på att förstärka eller efterlikna TGF‑beta3 specifikt i hjärtat, och därigenom stärka organets egen förmåga att begränsa skadlig ärrbildning samtidigt som nödvändig läkning i andra vävnader bevaras.
Citering: Xuan, J., Zhou, J., Huang, Y. et al. Cardiomyocyte-derived TGFB3 attenuates cardiac fibrosis and preserves cardiac function in heart failure. Sci Rep 16, 11534 (2026). https://doi.org/10.1038/s41598-026-42367-5
Nyckelord: hjärtsvikt, hjärtfibros, TGF‑beta3, kardiomyocyter, fibroblastsignalering