Clear Sky Science · de
Kardiomyozyten-abgeleitetes TGFB3 dämpft kardiale Fibrose und erhält die Herzfunktion bei Herzinsuffizienz
Warum das für Menschen mit geschwächtem Herzen wichtig ist
Herzinsuffizienz ist eine häufige und ernste Erkrankung, bei der das Herz nach und nach seine Pumpleistung verliert. Eine der Hauptursachen ist Vernarbung im Herzen, sogenannte Fibrose, die den Herzmuskel versteift und die Symptome verschlechtert. Diese Studie deckt ein überraschendes schützendes Signal auf, das von den Herzmuskelzellen selbst gebildet wird, das diesen Vernarbungsprozess verlangsamen und so die Herzfunktion erhalten kann. Das weist auf einen neuen, gezielteren Behandlungsansatz für Herzinsuffizienz hin.
Ein genauerer Blick auf Herzvernarbung
Bei Herzinsuffizienz „verschleißt“ das Herz nicht einfach. Vielmehr remodelt das Gewebe: Muskelzellen vergrößern sich oder gehen zugrunde, und Bindegewebszellen lagern überschüssiges Kollagen ein, sodass zähe narbenähnliche Bereiche entstehen. Seit Jahren steht eine mächtige Familie von Botenmolekülen, bekannt als TGF‑beta, in Verbindung mit dieser Vernarbung. Alle Formen von TGF‑beta zu blockieren erwies sich jedoch als zu grobe Methode, weil diese Moleküle auch bei normaler Reparatur und der Immunregulation eine Rolle spielen und das zu schweren Nebenwirkungen führte. Die neue Arbeit fragt, ob ein bestimmtes Familienmitglied, TGF‑beta3, dem Herz möglicherweise hilft, sich gegen außer Kontrolle geratene Fibrose zu verteidigen, statt sie zu fördern.
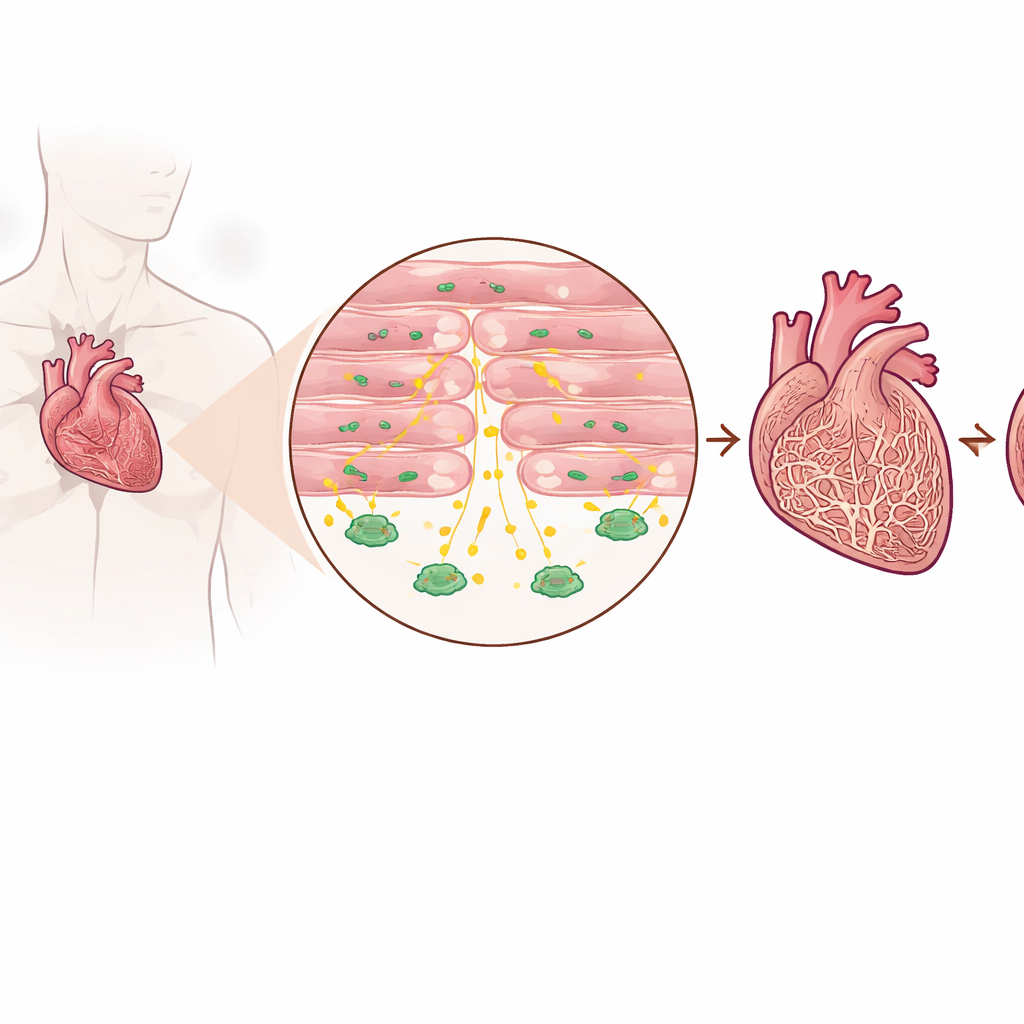
Ein schützendes Signal in kranken Herzen nachweisen
Die Forscher untersuchten Herzgewebe von Patientinnen und Patienten mit fortgeschrittener Herzinsuffizienz sowie von Personen ohne Herzinsuffizienz und nutzten mehrere Mausmodelle, die eine Druckbelastung des Herzens nachahmen. Sie fanden, dass die TGF‑beta3‑Spiegel in erkrankten Herzen beständig höher waren als in gesunden, sowohl im Gewebe selbst als auch im Blutkreislauf. Wichtig war, dass dieser Anstieg nicht hauptsächlich von den üblichen narbenbildenden Zellen ausging, sondern von Kardiomyozyten — den Muskelzellen, die tatsächlich kontrahieren, um Blut zu pumpen. Patienten mit höheren Blutspiegeln von TGF‑beta3 hatten tendenziell auch höhere Werte eines etablierten Herzinsuffizienzmarkers, was nahelegt, dass dieses Molekül die Intensität der Belastung widerspiegelt, der das Herz ausgesetzt ist.
Das Signal in Herzmuskelzellen ausschalten
Um zu prüfen, ob das von Kardiomyozyten produzierte TGF‑beta3 hilfreich oder schädlich ist, erzeugte das Team Mäuse, bei denen das Gen für TGF‑beta3 gezielt nur in Herzmuskelzellen gelöscht werden konnte. Unter normalen, ruhenden Bedingungen wiesen diese Mäuse Herzen auf, die in Aussehen und Funktion denen ihrer Wurfgeschwister sehr ähnelten. Wurden die Tiere jedoch einem Verfahren unterzogen, das die Hauptschlagader abdrückt — eine Standardmethode, um chronische Druckbelastung und letztlich Herzinsuffizienz zu induzieren — traten deutliche Unterschiede zutage. Mäuse ohne TGF‑beta3 in ihren Kardiomyozyten entwickelten eine schlechtere Pumpfunktion, größere Herzen und deutlich mehr Kollagenablagerungen zwischen den Muskelfasern als Kontrolltiere unter der gleichen Belastung.
Wie Herzmuskelzellen mit narbenbildenden Zellen kommunizieren
Bei der Untersuchung des Mechanismus verglichen die Forscher die Genaktivität in Herzen mit und ohne kardiales TGF‑beta3. Herzen ohne dieses Signal zeigten einen starken Anstieg zweier bekannter Treiber der Fibrose, CTGF und SERPINE1, die überwiegend von Fibroblasten, den Zellen der Bindegewebsbildung, produziert werden. In Zellkulturversuchen schalteten Fibroblasten, die dem klassischen Vernarbungssignal TGF‑beta1 ausgesetzt wurden, diese fibrotischen Gene ein und gingen in einen aktivierten, matrixproduzierenden Zustand über. Die Zugabe von TGF‑beta3 schwächte diese Reaktion jedoch ab. Fibroblasten, die mit Medium aus TGF‑beta3‑defizienten Kardiomyozyten behandelt wurden, wurden stärker aktiviert als solche, die Medium von normalen Kardiomyozyten erhielten — ein Hinweis darauf, dass die Muskelzellen normalerweise einen Faktor sezernieren, der eine Überaktivität der Fibroblasten einschränkt.
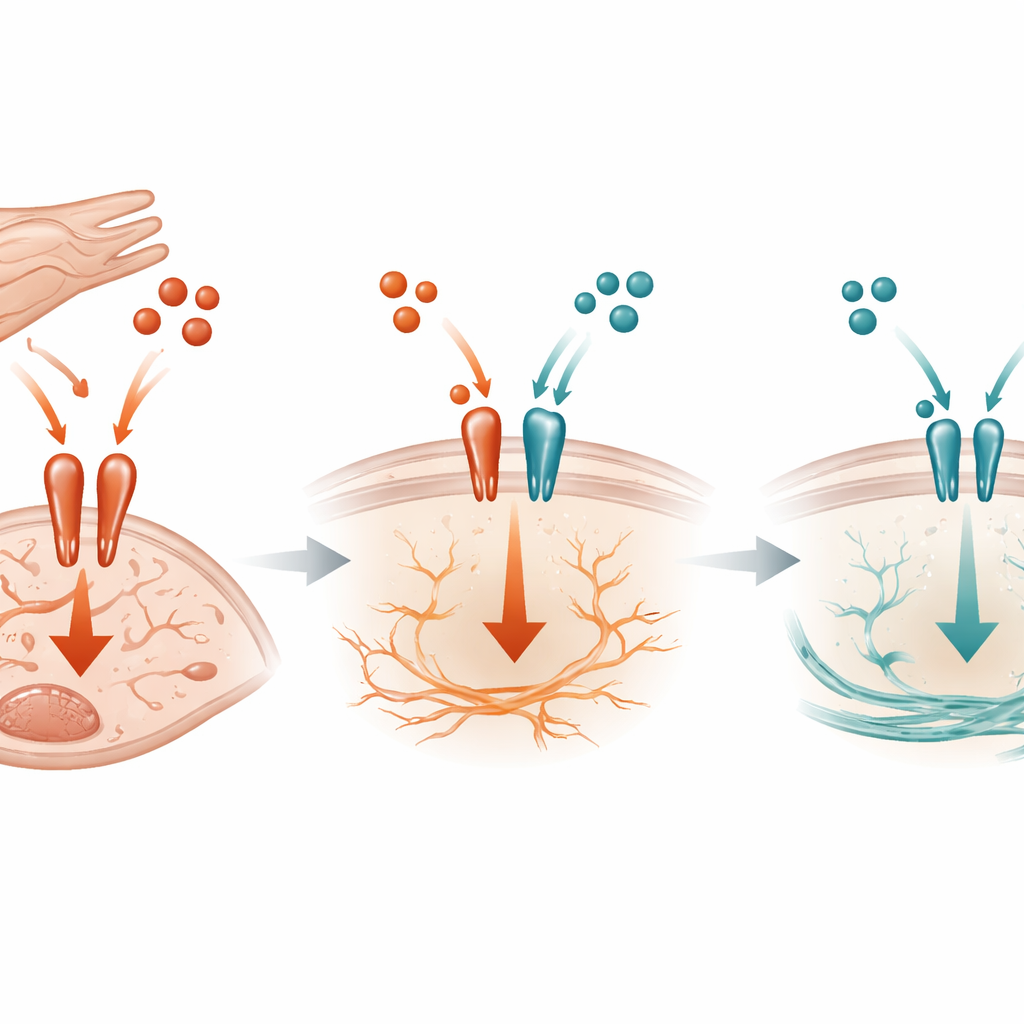
Ein molekularer Tauziehen an der Zelloberfläche
Auf molekularer Ebene zeigt die Studie, dass TGF‑beta3 wie eine konkurrierende Bremse gegenüber dem aggressiveren TGF‑beta1‑Signal wirkt. Beide Moleküle nutzen denselben Rezeptorkomplex an der Oberfläche von Fibroblasten, um im Inneren der Zelle Aktivität auszulösen. Biochemische Experimente ergaben, dass TGF‑beta3 an diese Rezeptoren binden und begrenzen kann, wie stark TGF‑beta1 andocken kann, wodurch die Aktivierung eines wichtigen Schalters im Inneren der Zelle, SMAD3, reduziert wird. Wenn kardiomyozytär abgeleitetes TGF‑beta3 bei Mäusen unter Druckbelastung fehlt, steigt die SMAD3‑Aktivierung, CTGF und SERPINE1 nehmen zu und die Fibrose beschleunigt sich. Bemerkenswert ist, dass dieser Effekt nicht von einem anderen bekannten Inhibitor innerhalb des Signalwegs abhängig war, was darauf hindeutet, dass die einfache Konkurrenz um den Rezeptor ein zentraler Bestandteil des Schutzmechanismus ist.
Was das für künftige Behandlungen bedeuten könnte
Für Nicht‑Spezialisten lautet die Kernbotschaft: Das versagende Herz ist nicht nur Opfer der Vernarbung; es versucht sich auch zu schützen. Herzmuskelzellen setzen TGF‑beta3 als lokale, eingebaute Abwehr frei, die gegen stärkere pro‑narbende Signale anzieht und verhindert, dass das Bindegewebe überreagiert. Fehlt dieses schützende Signal, verschlechtert sich die Fibrose und die Herzfunktion nimmt schneller ab. Statt das gesamte TGF‑beta‑System zu blockieren, könnten künftige Therapien darauf abzielen, TGF‑beta3 gezielt im Herzen zu steigern oder zu imitieren, um die eigene Fähigkeit des Organs zu stärken, schädliche Vernarbung zu begrenzen und gleichzeitig notwendige Heilungsprozesse in anderen Geweben zu erhalten.
Zitation: Xuan, J., Zhou, J., Huang, Y. et al. Cardiomyocyte-derived TGFB3 attenuates cardiac fibrosis and preserves cardiac function in heart failure. Sci Rep 16, 11534 (2026). https://doi.org/10.1038/s41598-026-42367-5
Schlüsselwörter: Herzinsuffizienz, kardiale Fibrose, TGF-beta3, Kardiomyozyten, Fibroblastensignalgebung