Clear Sky Science · fr
La TGFB3 d’origine cardiomyocytaire atténue la fibrose cardiaque et préserve la fonction cardiaque en cas d’insuffisance cardiaque
Pourquoi cela importe pour les personnes souffrant d’un cœur faible
L’insuffisance cardiaque est une affection fréquente et grave dans laquelle le cœur perd progressivement sa capacité à pomper le sang. Un coupable majeur est la formation de cicatrices à l’intérieur du cœur, appelée fibrose, qui rigidifie le muscle cardiaque et aggrave les symptômes. Cette étude met au jour un signal protecteur surprenant produit par les propres cellules musculaires du cœur, capable de ralentir ce processus de cicatrisation et d’aider à préserver la fonction cardiaque, ouvrant la voie à une approche thérapeutique nouvelle et plus ciblée pour traiter l’insuffisance cardiaque.
Un examen plus attentif de la cicatrisation cardiaque
Dans l’insuffisance cardiaque, le cœur ne se contente pas de « s’user ». Au contraire, ses tissus se remodèlent : les cellules musculaires grossissent ou meurent, et les cellules du tissu conjonctif déposent un excès de collagène, formant des zones rugueuses semblables à des cicatrices. Pendant des années, une famille puissante de molécules de signalisation connues sous le nom de TGF-bêta a été associée à cette fibrose. Bloquer toutes les formes de TGF-bêta s’est toutefois avéré trop large et a provoqué des effets secondaires graves, car ces molécules participent aussi à la réparation normale et à l’équilibre immunitaire. Le travail présenté ici pose la question de savoir si un membre particulier de cette famille, appelé TGF-bêta3, pourrait en réalité aider le cœur à se défendre contre une fibrose incontrôlée plutôt que de la favoriser.
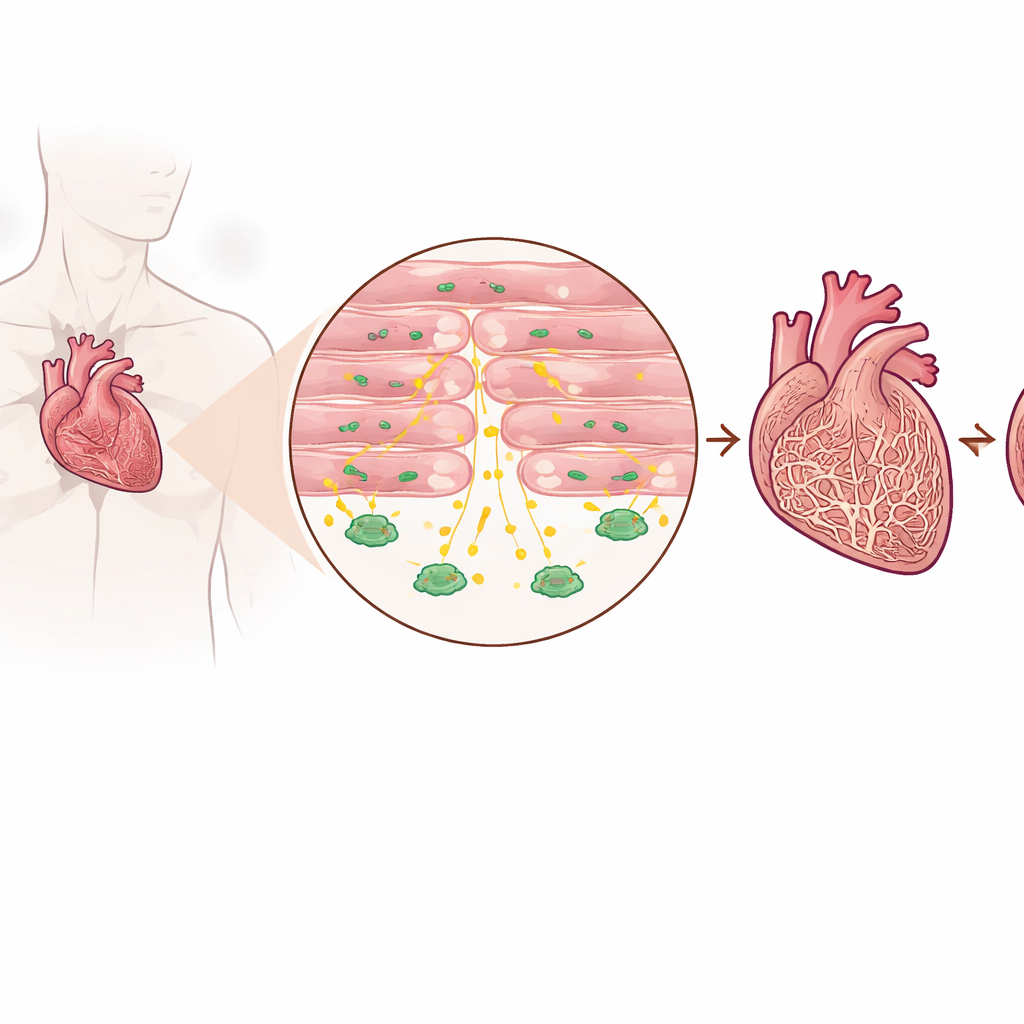
Détecter un signal protecteur dans les cœurs malades
Les chercheurs ont examiné des tissus cardiaques de patients atteints d’insuffisance cardiaque avancée et de personnes sans insuffisance, ainsi que plusieurs modèles murins reproduisant une surcharge de pression sur le cœur. Ils ont constaté que les niveaux de TGF-bêta3 étaient systématiquement plus élevés dans les cœurs défaillants que dans les cœurs sains, tant dans le tissu que dans le sang. Fait important, cette augmentation ne provenait pas principalement des cellules formant les cicatrices habituelles, mais des cardiomyocytes — les cellules musculaires qui se contractent pour pomper le sang. Les patients présentant des taux sanguins plus élevés de TGF-bêta3 avaient aussi tendance à présenter des niveaux plus élevés d’un marqueur standard d’insuffisance cardiaque, ce qui suggère que cette molécule reflète l’intensité du stress subi par le cœur.
Éteindre le signal dans les cellules musculaires cardiaques
Pour tester si ce TGF-bêta3 produit par les cardiomyocytes était bénéfique ou nuisible, l’équipe a conçu des souris chez lesquelles le gène codant pour TGF-bêta3 pouvait être supprimé sélectivement uniquement dans les cellules musculaires cardiaques. Dans des conditions normales et de repos, ces souris avaient des cœurs qui ressemblaient et fonctionnaient beaucoup comme ceux de leurs congénères. Mais lorsque les animaux ont été soumis à une intervention qui rétrécit l’artère principale quittant le cœur — une méthode standard pour induire une surcharge de pression chronique et une insuffisance cardiaque — les différences sont devenues frappantes. Les souris dépourvues de TGF-bêta3 dans leurs cardiomyocytes ont développé une fonction d’éjection plus faible, des cœurs plus volumineux et une accumulation de collagène entre les fibres musculaires bien plus importante que chez les animaux témoins exposés au même stress.
Comment les cellules musculaires cardiaques communiquent avec les cellules formant la cicatrice
Pour éclairer le mécanisme, les chercheurs ont comparé l’activité génique dans des cœurs avec et sans TGF-bêta3 cardiomyocytaire. Les cœurs privés de ce signal présentaient une forte augmentation de deux moteurs bien connus de la fibrose, CTGF et SERPINE1, produits principalement par les fibroblastes, les cellules responsables de la fabrication du tissu conjonctif. Dans des expériences en culture cellulaire, lorsque les fibroblastes étaient exposés au signal classique de cicatrisation TGF-bêta1, ils activaient ces gènes pro-fibrotiques et adoptaient un état activé producteur de matrice. L’ajout de TGF-bêta3 à ce contexte atténuait toutefois cette réponse. Les fibroblastes baignés dans le milieu provenant de cardiomyocytes dépourvus de TGF-bêta3 devenaient plus activés que ceux exposés au milieu de cardiomyocytes normaux, confirmant que les cellules musculaires sécrètent normalement un facteur qui freine la suractivité des fibroblastes.
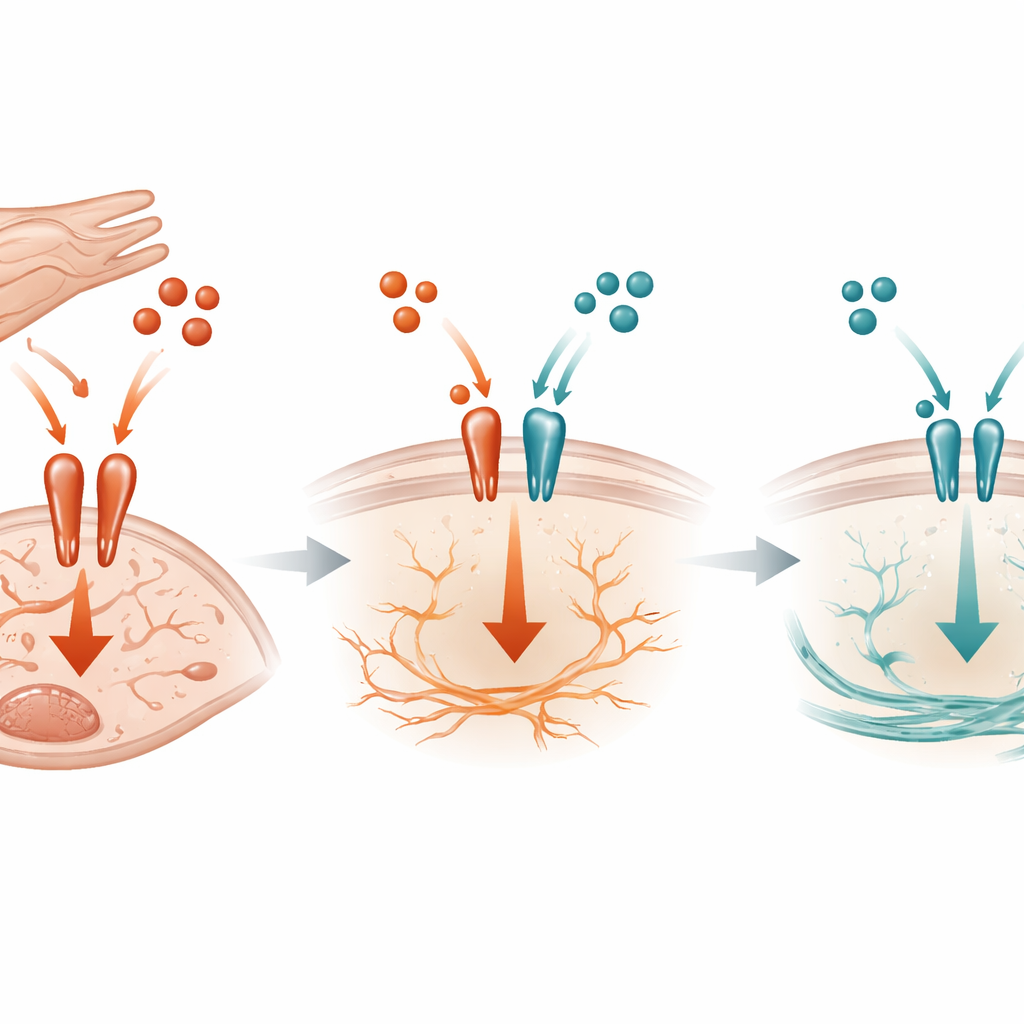
Une lutte moléculaire à la surface cellulaire
Au niveau moléculaire, l’étude montre que le TGF-bêta3 agit comme un frein compétitif sur le signal plus agressif TGF-bêta1. Les deux molécules utilisent le même complexe récepteur à la surface des fibroblastes pour déclencher l’activité à l’intérieur de la cellule. Des expériences biochimiques ont révélé que le TGF-bêta3 peut se lier à ces récepteurs et limiter la quantité de TGF-bêta1 qui peut s’y fixer, réduisant ainsi l’activation d’un commutateur clé intracellulaire appelé SMAD3. Lorsque le TGF-bêta3 d’origine cardiomyocytaire est absent chez des souris soumises à une surcharge de pression, l’activation de SMAD3 augmente, CTGF et SERPINE1 s’accroissent, et la fibrose s’accélère. Il est notable que cet effet ne dépendait pas d’un autre inhibiteur connu de la voie, ce qui suggère que la simple compétition pour le récepteur constitue une composante centrale du mécanisme protecteur.
Ce que cela pourrait signifier pour les traitements futurs
Pour un non-spécialiste, le message clé est que le cœur en insuffisance n’est pas seulement victime de la fibrose ; il tente aussi de se protéger. Les cellules musculaires cardiaques libèrent du TGF-bêta3 comme défense locale intégrée qui contrecarre des signaux pro-fibrotiques plus puissants et empêche le tissu conjonctif de basculer dans une hyperactivité néfaste. Quand ce signal protecteur manque, la fibrose s’aggrave et la fonction cardiaque décline plus rapidement. Plutôt que de bloquer l’ensemble du système TGF-bêta, les thérapies futures pourraient viser à renforcer ou imiter spécifiquement le TGF-bêta3 au niveau cardiaque, renforçant la capacité intrinsèque de l’organe à limiter les cicatrices nuisibles tout en préservant la réparation nécessaire dans d’autres tissus.
Citation: Xuan, J., Zhou, J., Huang, Y. et al. Cardiomyocyte-derived TGFB3 attenuates cardiac fibrosis and preserves cardiac function in heart failure. Sci Rep 16, 11534 (2026). https://doi.org/10.1038/s41598-026-42367-5
Mots-clés: insuffisance cardiaque, fibrose cardiaque, TGF-bêta3, cardiomyocytes, signalisation des fibroblastes