Clear Sky Science · pt
TGFB3 derivado de cardiomiócitos atenua a fibrose cardíaca e preserva a função cardíaca na insuficiência cardíaca
Por que isso importa para pessoas com corações fracos
A insuficiência cardíaca é uma condição comum e grave em que o coração perde gradualmente sua capacidade de bombear sangue. Um grande culpado é a formação de cicatriz dentro do coração, chamada fibrose, que torna o músculo cardíaco mais rígido e piora os sintomas. Este estudo revela um sinal protetor surpreendente produzido pelas próprias células musculares do coração que pode retardar esse processo de cicatrização e ajudar a preservar a função cardíaca, apontando para uma nova forma mais precisa de tratar a insuficiência cardíaca.
Um olhar mais atento sobre a cicatrização cardíaca
Na insuficiência cardíaca, o coração não simplesmente “se desgasta”. Em vez disso, seu tecido se remodela: as células musculares aumentam de tamanho ou morrem, e as células do tecido conjuntivo depositam colágeno em excesso, formando áreas duras semelhantes a cicatrizes. Por anos, uma família poderosa de moléculas de sinalização conhecida como TGF-beta tem sido associada a essa cicatrização. Bloquear todas as formas de TGF-beta, no entanto, revelou-se um instrumento impreciso demais, causando efeitos colaterais sérios porque essas moléculas também ajudam na reparação normal e no equilíbrio imunológico. O trabalho novo questiona se um membro específico dessa família, chamado TGF-beta3, pode na verdade ajudar o coração a se defender contra a fibrose descontrolada em vez de promovê-la.
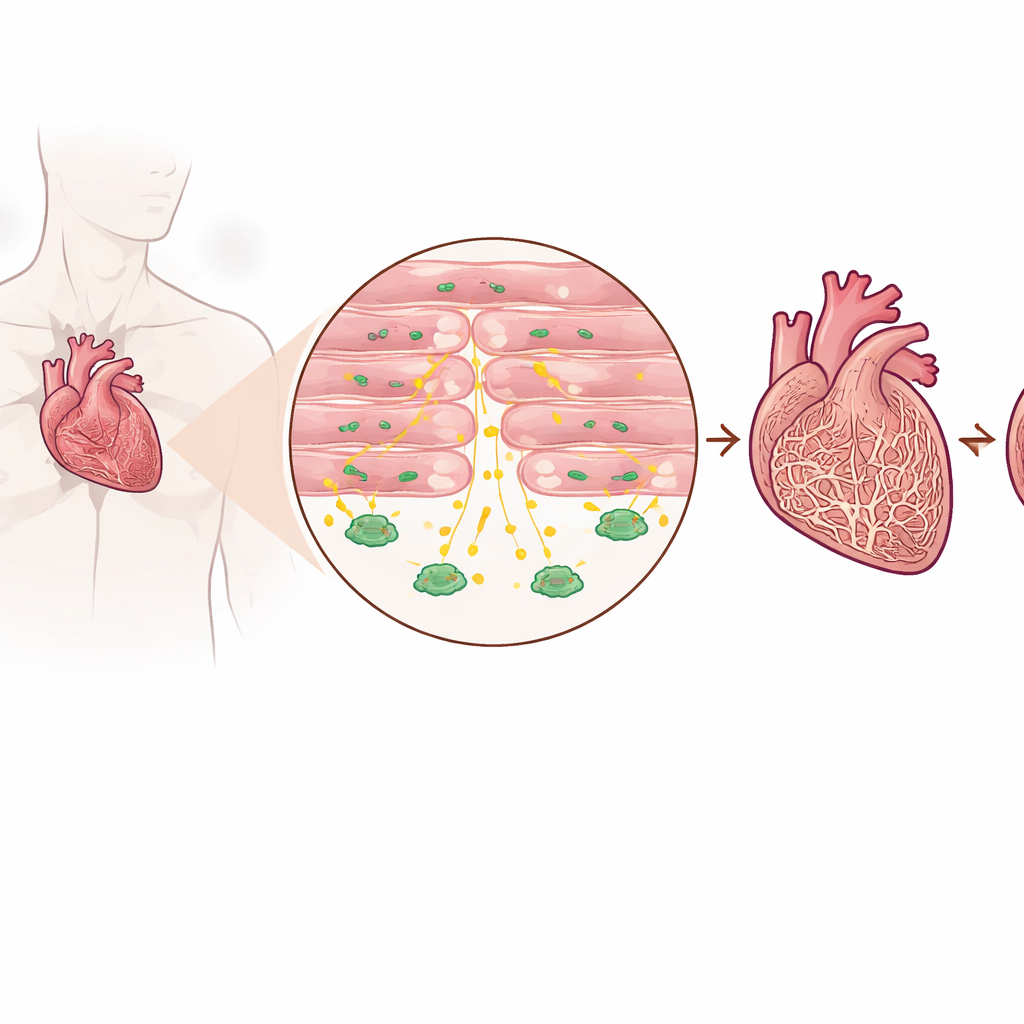
Detectando um sinal protetor em corações doentes
Os pesquisadores examinaram tecido cardíaco de pacientes com insuficiência cardíaca avançada e de pessoas sem insuficiência, além de vários modelos de camundongos que mimetizam sobrecarga de pressão no coração. Eles descobriram que os níveis de TGF-beta3 eram consistentemente mais altos em corações em falha do que em corações saudáveis, tanto no próprio tecido quanto na corrente sanguínea. Importante, esse aumento não provinha principalmente das usuais células formadoras de cicatriz, mas dos cardiomiócitos — as células musculares que realmente se contraem para bombear o sangue. Pacientes com níveis sanguíneos mais altos de TGF-beta3 também tendiam a ter níveis maiores de um marcador padrão de insuficiência cardíaca, sugerindo que essa molécula reflete a intensidade do estresse ao qual o coração está submetido.
Desligando o sinal nas células musculares do coração
Para testar se o TGF-beta3 produzido pelos cardiomiócitos era útil ou prejudicial, a equipe engenheirou camundongos nos quais o gene do TGF-beta3 podia ser excluído seletivamente apenas nas células musculares cardíacas. Em condições normais e em repouso, esses camundongos apresentavam corações que se pareciam e funcionavam de modo muito semelhante aos de seus irmãos de ninhada. Mas quando os animais foram submetidos a um procedimento que estreita a principal artéria que sai do coração — uma forma padrão de induzir sobrecarga crônica de pressão e eventual insuficiência cardíaca — as diferenças tornaram-se marcantes. Camundongos sem TGF-beta3 em seus cardiomiócitos desenvolveram função de bombeamento pior, corações maiores e muito mais acúmulo de colágeno entre as fibras musculares do que animais controle expostos ao mesmo estresse.
Como as células musculares do coração se comunicam com as formadoras de cicatriz
Investigando o mecanismo, os pesquisadores compararam a atividade gênica em corações com e sem TGF-beta3 nos cardiomiócitos. Corações sem esse sinal mostraram um aumento acentuado em dois conhecidos impulsionadores da fibrose, CTGF e SERPINE1, que são produzidos principalmente por fibroblastos, as células responsáveis por fabricar o tecido conjuntivo. Em experimentos em cultura celular, quando fibroblastos foram expostos ao clássico sinal de cicatrização TGF-beta1, eles ligaram esses genes fibróticos e adotaram um estado ativado, produtor de matriz. Adicionar TGF-beta3 à mistura, porém, atenuou essa resposta. Fibroblastos banhados por fluido de cardiomiócitos deficientes em TGF-beta3 tornaram-se mais ativados do que os expostos ao fluido de cardiomiócitos normais, confirmando que as células musculares normalmente secretam um fator que restringe a hiperatividade dos fibroblastos.
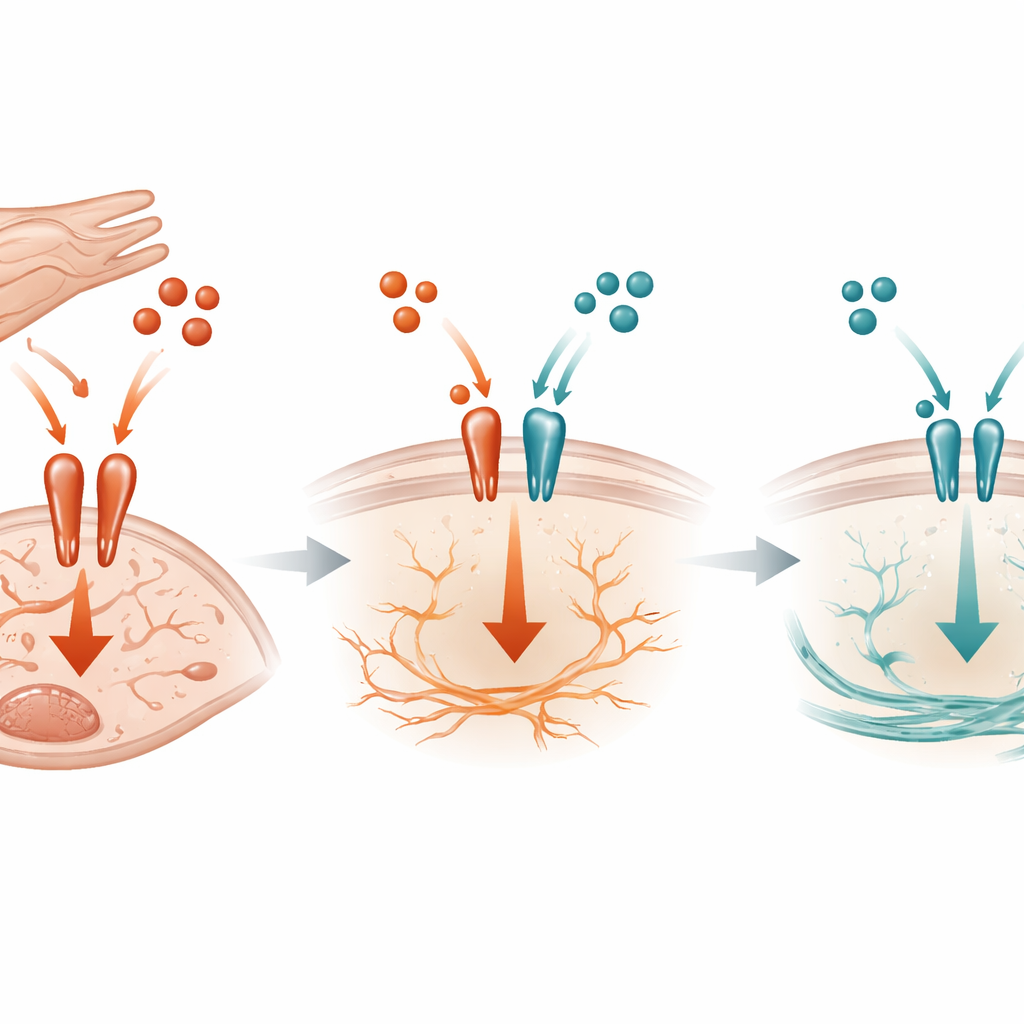
Uma disputa molecular na superfície celular
Ao nível molecular, o estudo mostra que o TGF-beta3 age como um freio competitivo sobre o sinal mais agressivo do TGF-beta1. Ambas as moléculas usam o mesmo complexo de receptores na superfície dos fibroblastos para desencadear atividade dentro da célula. Experimentos bioquímicos revelaram que o TGF-beta3 pode se ligar a esses receptores e limitar quanto o TGF-beta1 pode se acoplar, reduzindo assim a ativação de um interruptor chave dentro da célula chamado SMAD3. Quando o TGF-beta3 derivado de cardiomiócitos está ausente em camundongos sob sobrecarga de pressão, a ativação de SMAD3 aumenta, CTGF e SERPINE1 crescem, e a fibrose acelera. Notavelmente, esse efeito não dependia de outro inibidor conhecido dentro da via, sugerindo que a simples competição pelo receptor é parte central do mecanismo protetor.
O que isso pode significar para tratamentos futuros
Para um não especialista, a mensagem chave é que o coração em falha não é apenas uma vítima da cicatrização; ele também tenta se proteger. As células musculares cardíacas liberam TGF-beta3 como uma defesa local e embutida que puxa contra sinais pró-cicatrização mais fortes e impede que o tecido conjuntivo entre em excesso. Quando esse sinal protetor está ausente, a fibrose piora e a função cardíaca declina mais rapidamente. Em vez de bloquear todo o sistema TGF-beta, terapias futuras podem visar aumentar ou imitar especificamente o TGF-beta3 no coração, potencializando a capacidade do órgão de limitar a cicatrização prejudicial enquanto preserva a cura necessária em outros tecidos.
Citação: Xuan, J., Zhou, J., Huang, Y. et al. Cardiomyocyte-derived TGFB3 attenuates cardiac fibrosis and preserves cardiac function in heart failure. Sci Rep 16, 11534 (2026). https://doi.org/10.1038/s41598-026-42367-5
Palavras-chave: insuficiência cardíaca, fibrose cardíaca, TGF-beta3, cardiomiócitos, sinalização de fibroblastos